罗飞宏教授:人类遗传病的分类和相关疾病
遗传缺陷或异常是包括肿瘤、糖尿病等众多慢性疾病发病的共同机制,随着传染性疾病的控制,遗传病已成为人类致残、致死的主要原因之一。遗传病种类繁多,大多数人类遗传缺陷可归类为染色体病、单基因孟德尔遗传病、单基因非孟德尔遗传病、多基因遗传病等。目前可供查询的常染色体、X连锁、Y连锁、线粒体类等疾病或关联表型已超过2万种。
一、染色体病
【染色体核型】
正常人体细胞染色体共23对,1~3号染色体为A组,4~5染色体为B组,6~12+X染色体为C组,13~15号染色体为D组,16~18号染色体为E组包括,19~20号染色体为F组,21~22+Y染色体为G组。染色体异常所引起的疾病主要分为染色体数目异常、结构异常和性染色体异常三类。为判别染色体异常与否,至少需要观察20个细胞染色体分裂象,20个细胞中发现3个相同数目减少或2个细胞有相同发现,可判别为异常;如果20个细胞仅发现1个异常染色体分裂象,则需增加观察30个细胞。染色体结构畸变的表示方法有简式和详式两种,以临床常用的简式表达如 46,XY,del(2)(q31q35)为例,依次为染色体总数、性染色体组成、畸变类型的符号、括号内为受累染色体序号、在接着的括号内为受累染色体断裂点(其他染色体畸变符号见表1)。
表1 染色体结构畸变的表示方法
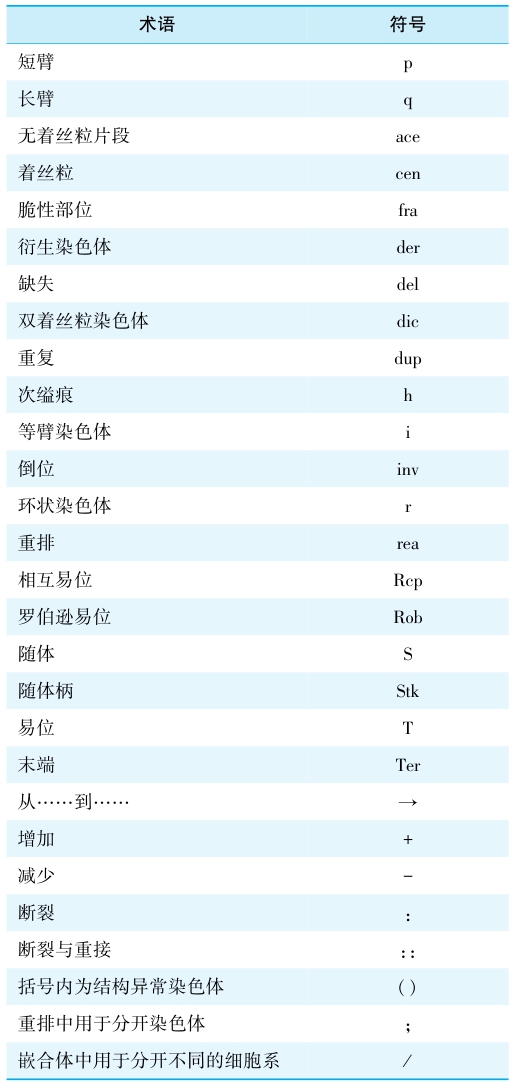
【染色体病的病因】
(一)染色体数目异常
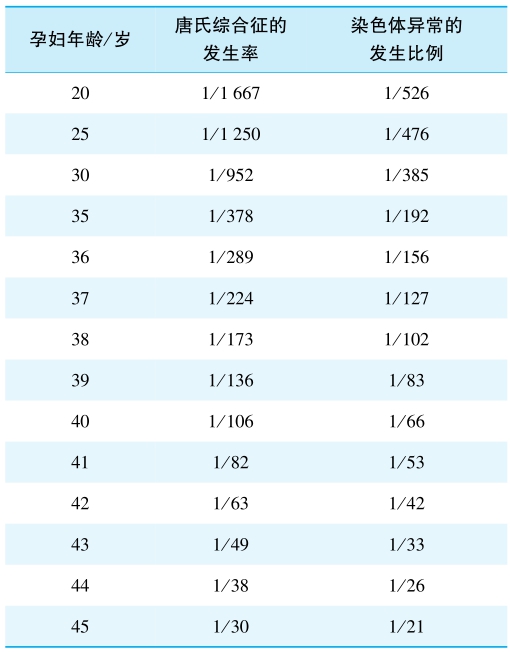
染色体数目异常可为整倍体(euploid)、非整倍体(aneuploidy)、嵌合体(chimaera)三种。 数目成倍增加或者减少的称为整倍体异常。数目成单个或几个增加或减少的称为非整倍体,非整倍体产生于生殖细胞减数分裂过程中,染色体不分离和减数分裂时染色体丢失,包括单体、缺体、三体、多体。染色体的嵌合体产生于受精卵卵裂染色体不分离和受精卵卵裂染色体丢失,患者体内同时二倍体和非整倍体。胎儿染色体异常的发生危险度与孕妇的年龄间存在密切关系(表2)。
表2 孕妇年龄与染色体异常发生间的关系
(二)染色体结构畸变
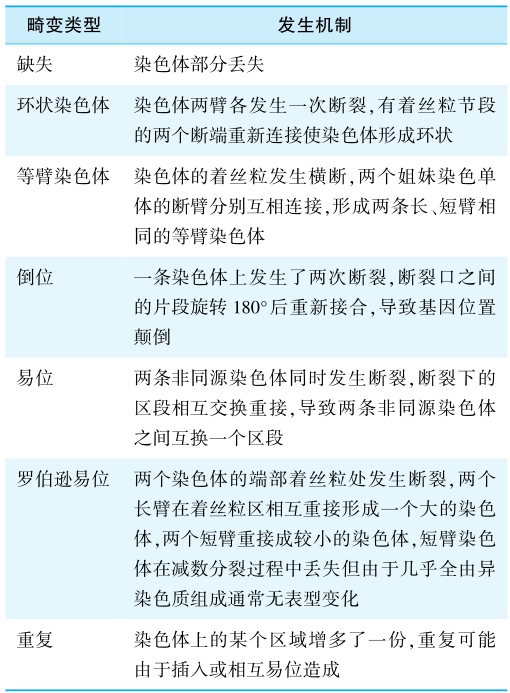
染色体结构畸变常见的有缺失、环状染色体、等臂染色体、倒位、易位、罗伯逊易位、重复、平衡易位携带者等(表3)。
表3 染色体结构畸变的类型及发生机制
【人类染色体病的分类】
(一)常染色体病
由于1~22号常染色体先天性数目异常所致的遗传病症称为常染色数目异常体病,常分为三体综合征、单体综合征、部分三体综合征和部分单体综合征。此类患者通常症状严重,可出现先天性多发畸形、特殊体貌肤纹,一般均伴有较严重或明显的生长与智力发育落后。常见的染色体病见表4。
表4 常见的三体型常染色体遗传病
(二)性染色体病
由于性染色体X或Y染色体结构或数目异常引起的疾病,常见的为Turner综合征、X三体综合征、克氏综合征、47,XYY综合征等。
与Turner综合征的X染色体缺失临床症状明显不同,Y染色体异常的疾病临床症状较为轻微,可能与Y染色体携带相对较少的基因有关。在女性胚胎体细胞发育早期(精子、卵子除外)一条X染色体发生化学修饰,被修饰的基因不再转录表达,这个过程被称为X染色体失活(X chromosome inactivation)。X染色体失活在胚胎多数体细胞中是随机性发生的,大约一半的体细胞组织灭活母系来源的X染色体,另一半将灭活父系来源的X染色体。X染色体失活的现象可以防止携带在两个拷贝的X染色体的女性基因被表达两次,而男性由于只有一条X染色体只能表达一次。如果细胞定向生成卵子,则无X染色体失活现象。灭活的X染色体通常复制晚于其他染色体,并浓缩形成巴尔小体。
正常的女性由于X染色体的失活,她们的部分体细胞只表达母源性X染色体的基因,而其他体细胞只表达父源性X染色体的基因,父源和母源失活基因的比例并不是精确的1∶1比例,X染色体随机失活的比例是可变的,也并非在X染色体上的所有基因都失活,少量基因可逃逸修饰并在两个X染色体中都表达。X染色体的缺失或过多所出现的临床表型异常与上述机制有关。
克兰费尔特综合征[Klinefelter syndrome(47,XXY)]发生于男性,与正常女性类似其体细胞中的两个X染色体中的一个会随机失活,因此其临床表型相对与缺少一条 X染色体的Turner综合征轻,表现高身材和不育症,可出现男性乳房发育。X三体综合征[trisomy X syndrome(47,XXX)]女性虽然会出现月经不规则或不育,三个X染色体中的两个会被灭活,临床表现上总体良性。47,XYY综合征男性表现为高身材,偶尔可见隐睾,睾丸发育不全并有精子形成障碍和生育力下降,尿道下裂等,但大多数男性可以生育,少数有轻度学习障碍。48,XXXY或49,XXXXY人极为罕见,临床表现出类似于克氏综合征患者,但严重程度稍微增加。
二、单基因孟德尔遗传病
19世纪60年代孟德尔从豌豆的研究中发现了分离定律和自由组合定律,统称为孟德尔遗传规律,有关的疾病通常被归类为常染色体显性遗传、常染色体隐性遗传或性连锁遗传。
(一)常染色体显性遗传
假设正常亲代基因为A,突变基因为a,子代的基因型为杂合子Aa,若杂合子Aa能表现出与显性基因a有关的性状或遗传病时,其遗传方式称为显性遗传。常染色体显性遗传(autosomal dominant inheritance,AD)是指控制性状或疾病的显性基因位于常染色体的遗传方式。目前已发现超过2 000多种疾病与AD遗传模式有关,发病特点为患者双亲中有一方同为患者,家系中连续几代均有患者,患者的同胞中约有1/2是患者,发病男女机会均等,如果双亲均不是患者,则子女一般也不发病。随环境的变化,杂合子的外显率也受特定环境因素的影响(表5)。
表5 常染色体显性遗传的不同遗传类型

(二)常染色体隐性遗传
遗传性状或病症关联基因位于常染色体上,但在杂合状态(Aa,A为正常基因)下不表现相应性状或发病,只有纯合体(aa)才表现,称为常染色体隐性遗传(autosomal recessive inheritance,AR)。AR模式特点有致病基因位于常染色体,遗传与性别无关,不一定每代都出现患者;患者双亲表现型可以正常;患者与外表上正常的异性结婚,其子女可能患病,也可能全正常;若患者与表现型正常而基因型不正常(基因型为Aa)的异性结婚,则其子女中患病概率和为致病基因携带者的可能性均为50%,近亲婚配时,子女患病的机会较多。
目前已报道近2 000余种性状呈现AR模式,如白化病、镰状细胞贫血、苯丙酮尿症等。早期进行基因检测明确诊断,有可能预防此类疾病损害的发生,如苯丙氨酸羟化酶(phenylalanine hydroxylase,PAH)能催化苯丙氨酸转化为酪氨酸,PAH基因突变导致体内苯丙氨酸堆积,部分苯丙氨酸通过旁路途径被转化为苯丙酮酸,婴幼儿血液和其他组织中堆积了高浓度的苯丙酮酸和未转化的苯丙氨酸,导致智力低下,早期基因诊断明确后通过饮食调整可以有效防止智力损害的发生。
(三)X连锁遗传
位于X染色体上的基因控制性状的遗传即是X连锁遗传(X-linked inheritance),对于X连锁基因来说,女性是纯合子或杂合子,男性是半合子。女性的两条X染色体一条来自母方,另一条来自父方,并随机地一条传给女儿、另一条传给儿子;男性的一条X染色体始终只能来自母方,且只能传给女儿而不能传给儿子,因此X连锁遗传的最大特点是性状或遗传病不存在从男性到男性的传递。
1.X连锁隐性遗传
遗传病或特定性状有关的遗传基因位于X染色体上,如果该基因是隐性的,其遗传方式称为X连锁隐性遗传(X-linked recessive inheritance,XR)。在女性中,当X连锁隐性基因处于杂合状态(XAXa,A为正常基因)时,个体不发病;只有当两条X染色体上的处于基因纯合(XaXa)状态才发病;在男性中,只要X染色体上有隐性致病基因(XaY)就会发病,因此在自然人群中XR男性的发病率比女性高很多,家系中往往只看到男性发病。如红绿色盲我国人群中男性发病率为7%,女性发病率只有0.5%。
2.X连锁显性遗传
特定遗传性状或遗传病的基因位于X染色体上,如果基因表现型是显性的,该遗传方式为X连锁显性遗传(X-linked dominant inheritance,XD)。XD不论男性(XaY)还是女性(XaXa,XAXa),只要携带致病基因(a)均会发病,由于女性比男性多一条X染色体,因此自然人群中女性的发病率显著高于男性。
(四)Y连锁遗传
人类Y染色体仅存在于男性个体,Y染色体上的基因决定的性状只能由父亲传给儿子,不传给女儿,所以呈限雄遗传(holandric inheritance)。Y染色体上的基因很少,大多数基因在X染色体上没有相应的等位基因,基因所控制的性状只能在男性中表现出来,在家族中以男性-男性的方式遗传。
(五)从性遗传
如果常染色体上基因所控制的性状,在表型上受性别影响,发病呈现男女性分布比例或表现程度上的差异,这种遗传方式称为从性遗传(sex-influenced inheritance)。如遗传性早秃为常染色体遗传病,基因在男性为显性,杂合子男性即会出现早秃性状;在女性为隐性,女性杂合子不出现早秃性状,只有纯合体才会表现早秃症状,症状也比男性轻。
(六)限性遗传
某种性状或遗传病的基因位于常染色体或性染色体上,不论显性还是隐性遗传,只在一种性别中得以表现,而另一性别中完全不能表现,基因的性状可以后代传递,这种遗传方式称为限性遗传(sex-limited inheritance)。如前列腺癌关联基因在常染色体,但只有男性患病,女性不患病。
三、单基因非孟德尔遗传病
许多单基因疾病并非遵循孟德尔遗传模式,称为非孟德尔遗传(non-Mendelian inheritance),非孟德尔遗传病的发病机制也越来越多地被发现,相当多的肿瘤、综合征、智力低下、精神发育异常、躯体发育畸形等与非孟德尔遗传有密切关系。
(一)三核苷酸重复疾病
三核苷酸重复序列是指三个不同的碱基为一个单位重复排列而形成的DNA序列,如(CTG)n、(CAG)n、(CGG)n、(CCG)n 等。 三核苷酸重复疾病(trinucleotide repeat disease,TRD)或三核苷酸扩增性疾病(trinucleotide expansion disease,TED)是指一类由于致病基因内部或调控区域三核苷酸重复序列拷贝数目的不稳定而异常扩增导致的一类疾病。基因组DNA上三核苷酸重复序列从一代向下一代传递时往往存在进一步扩增趋势,这种拷贝数不稳定地异常扩展现象即动态突变(dynamic或expansion mutation)。
三核苷酸重复序扩增可以发生在DNA的任何位置,可在基因内或外、基因的外显子内或内含子中,也可在基因的编码区或非编码区中。如三核苷酸重复序列位于编码区,可以导致编码的蛋白质产物长度增加,如亨廷顿病;如三核苷酸重复发生在非编码区,非编码区域延长时,会干扰基因产物的正确表达,如脆性X综合征(FMR-1基因5′端非翻译区CGG重复,正常人CGG拷贝数为2~54;患者大于200)。
TRD在代代相传的过程中,发病年龄逐步提前,称为早现现象。早现现象与重复的次数相关,从一代传递到下一代过程汇中重复的次数不断扩大导致越来越不稳定,直到发生“全突变”,达到全突变后其遗传方式遵循标准孟德尔方式。
(二)线粒体DNA突变
线粒体是人类细胞中除细胞核外唯一具有自身遗传物质的细胞器。线粒体基因组mtDNA分子以多拷贝的形式存在于线粒体和细胞中,分别编码2个ATP合成酶亚基、1个细胞色素b亚基(Cytb)、3个细胞色素c氧化酶亚基、7个NADH-泛醌还原酶ND亚基、2个rRNAs和22个tRNAs。线粒体DNA突变遗传病特点如下:
1.母系遗传(maternal inheritance)
在受精过程中,精子线粒体会被卵子中泛素水解酶特异性识别而降解,导致只有母亲能将其mtDNA分子传递给下一代,再通过女儿传播给后代。
2.异质性和突变负荷
线粒体基因突变可以发生在成千上万个mtDNA分子上,突变产生的mtDNA突变体含量介于0~100%之间,人体组织或细胞可同时拥有突变型和野生型mtDNA的现象称为异质性(heteroplasmy);组织或细胞仅有同一种mtDNA(全部为突变型或全部为野生型mtDNA)的现象称为均质性(homoplasmy)。突变负荷(mutation load)是指发生突变的mtDNA占全体mtDNA的百分比,mtDNA疾病的发生及其临床表型往往取决于突变体突变负荷的高低。
3.阈值效应
当突变负荷超过一定程度后,野生型mtDNA代偿不足,组织或器官就会出现异常,称为阈值效应(threshold effect)。能量需求高的部位(如脑、心、骨骼肌、内分泌腺等)容易受突变影响,较低的突变负荷就能引起临床症状;能量需求低的部位(如肺、皮肤等)需较高的突变负荷才能引发疾病。
4.“瓶颈”和随机分配
异质性mtDNA突变体的突变负荷高低在不同的世代交替间变化显著,这种效应即为线粒体遗传的“瓶颈”。“瓶颈”现象机制不清,可能与卵母细胞经历了多次分裂使得最终分配到每个卵子中的mtDNA的有效数量较少所致。体细胞包括卵细胞每经历一次有丝分裂,mtDNA会随着线粒体一起被随机分配到子代细胞中,组织中mtDNA的突变负荷会随组织细胞分裂而变化,同一患者的疾病表型也会随时间推移而表现出变异性,造成同一母系家族成员间的疾病表型和同一患者组织间的突变负荷时常会迥然不同。
(三)表观遗传(epigenetics)
指在DNA序列没有变化的情况下,生物的表型发生了可遗传的改变,即可遗传的基因组表观修饰。肿瘤、孤独症、智力和精神遗传,以及综合征如Prader-Willi综合征、Angelman综合征等的发病均与表观遗传有关;除X染色体失活、非编码RNA调控外,表观遗传关联疾病的发生机制主要有:
1.DNA甲基化
DNA甲基化是指在DNA甲基转移酶(DNMTs)的催化下,将甲基基团转移到胞嘧啶碱基上的一种修饰方式。DNA高度甲基化首先会影响DNA结构,进而阻遏基因转录,引起基因沉默。
2.组蛋白修饰
组蛋白的修饰包括乙酰化、甲基化、磷酸化、泛素化、腺苷酸化、甲基化等,上述修饰方式可以阻遏也可以促进基因的转录。组蛋白乙酰化标志着其处于转录活性状态;组蛋白低乙酰化或去乙酰化表明处于非转录活性状态。组蛋白乙酰化的异常可以导致Rett综合征、肿瘤等疾病的发生。
3.染色质重塑
染色质重塑是DNA甲基化、组蛋白修饰、染色质重塑复合物共同作用于影响核小体结构,为其他蛋白提供和DNA的结合位点。染色质重塑异常,可导致多种综合征如Cockayne综合征、Schimke综合征、智力落后及肿瘤。
4.基因组印记
分别来自源于父源和母源的等位基因,因表观遗传修饰而出现差异性表达,其中一个可以表达,而另一个沉默不表达的现象称基因组印记(genomic imprinting)。人体中已知有80多种印记基因,印记丢失导致等位基因同时表达或有活性的等位基因突变,均可引起人类疾病。如新生儿糖尿病、Beckwith-Weideman综合征也与印记基因异常有关。
四、多基因遗传病
多基因遗传病(multifactorial inheritance disorders)是指由多对微效基因和环境因素共同作用而引起的遗传病。多基因遗传病属于数量性状遗传病,单个基因并不致病,多个基因的作用累积共同参与发病过程的形成,存在阈值现象和性别差异。精神分裂症、哮喘、高血压、冠状动脉粥样硬化等100多种常见疾病为多基因遗传模式。多基因遗传病发病呈家族聚集倾向,患者一级亲属的发病率高于群体发病率,但家族成员中发病率比单基因遗传病发病率低,如肥胖症、2型糖尿病的遗传特点。多基因遗传病产生于多对缺陷基因作用的累积效应,家族致病基因含量越多,家族成员发病风险越高,发病的人数越多,患者病情越严重。不同民族、种族遗传基因存在差异,导致疾病具有种族特异性或聚集性,如太平洋岛国瑙鲁、美国土著Pima印第安人的2型糖尿病发病率分别高达30%和50%。发病受遗传因素和环境因素的双重影响,遗传率高说明遗传基因影响大、环境因素影响小,遗传率低环境因素影响大、遗传基因影响小。亲缘关系越近,发病风险越高;亲缘关系越低,发病风险越低。如发病存在性别差异,发病率越低的性别,导致发病的基因阈值就越高,携带的基因可能就越多,其子代发病风险就越高。
遗传病的发生机制复杂,随着DNA测序技术、基因调控机制研究的进展,不断有新的致病基因和遗传调控机制被发现出来,为遗传关联疾病的治疗带来新的希望和研究方向。
推荐阅读
[1]贺林,马端,段涛,等.临床遗传学[M].上海:上海科学技术出版社,2013.
[2]RIEGEL M.Human molecular cytogenetics:From cells to nucleotides[J].Genet Mol Biol,2014,37(1):194-209.
[3]HARVEY Z H,CHEN Y,JAROSZ D F.Protein-Based Inheritance:Epigenetics beyond the Chromosome[J].Mol Cell,2018,69(2):195-202.
[4]DEN DUNNEN W F A.Trinucleotide repeat disorders[M].The Netherlands:Springer,2017:383-391.
知识来源
来源:人卫知识数字服务体系
作者:罗飞宏教授,复旦大学附属儿科医院内分泌遗传代谢科
专家简介
罗飞宏,复旦大学附属儿科医院内分泌遗传代谢科主任,主任医师,博士生导师,医学博士。中华医学会儿科分会儿科内分泌遗传代谢病学组副组长,上海医学会儿科分会儿科内分泌遗传代谢病学组副组长,上海医学会儿科分会委员,中华医学会糖尿病分会1型糖尿病学组委员,亚太儿科内分泌协会、欧洲内分泌协会、国际青少年糖尿病联盟成员。主持有国家自然科学基金、上海市科委基金等科研项目,参与国家十一五、十二五科技攻关项目多项。主要从事儿科内分泌遗传代谢性疾病的临床诊治和发病机制研究,对儿童糖尿病、矮小症、性早熟和有机酸血症等内分泌遗传代谢疾病的诊断和处理有较系统的临床诊治经验,对一些新型方法如干细胞移植等有一定的经验积累。
- 评价此内容
- 我要打分




 会员登录
会员登录
近期推荐
- 张丹副教授:ICU获得性肌无力…
- 许媛教授:重症营养支持国际指…
- 李维勤副主委:从指南变迁看重…
- 戴体俊主委:“中药麻醉”的回…
- 王昱副主委:移植后恶性血液病…
- 刘开彦副所长:血型不合的造血…
- 廖二元教授:简述能量代谢、物…
- 罗飞宏教授:人类遗传病的分类…
- 支玉香教授:过敏性疾病详解
- 画伟教授:艾滋病常见机会性感…

热点文章
- 还没有任何项目!
热门关键词
最新会议
- 2013循证医学和实效研究方法学研讨会
- 欧洲心脏病学会年会
- 世界帕金森病和相关疾病2013年会议
- 英国介入放射学学会2013年第25届年会
- 美国血液学会2013年年会
- 美国癫痫学会2013年第67届年会
- 肥胖学会 2013年年会
- 2013年第9届欧洲抗体会议
- 国际精神病学协会 2013年会议
- 妇科肿瘤2013年第18届大会
- 国际创伤压力研究学会2013年第29届…
- 2013年第4届亚太地区骨质疏松症会议
- 皮肤病协会国际2013年会议
- 世界糖尿病2013年大会
- 2013年国际成瘾性药年会
- 彭晓霞---诊断试验的Meta分析
- 武姗姗---累积Meta分析和TSA分析
- 孙凤---Network Meta分析
- 杨智荣---Cochrane综述实战经验分享
- 杨祖耀---疾病频率资料的Meta分析
友情链接
合作伙伴
Copyright g-medon.com All Rights Reserved 环球医学资讯 未经授权请勿转载!
网络实名:环球医学:京ICP备08004413号-2
关于我们|
我们的服务|版权及责任声明|联系我们
互联网药品信息服务资格证书(京)-经营性-2017-0027
互联网医疗保健信息服务复核同意书 京卫计网审[2015]第0344号
