龚凤英:基因突变的几种常见检测技术
遗传性内分泌基因病,包括单基因病和多基因病,其基因诊断则选择不同基因突变检测技术,直接检测和分析样本中的DNA或RNA水平上的致病性突变或者变异,常见的DNA变异检测技术见表1。样本只要包含任何有核细胞即可,包括外周血(最常用)、活检组织标本、手术切除标本等等。
一、直接检测DNA突变的技术
(一)聚合酶链反应技术
聚合酶链反应(polymerase chain reaction,PCR)是最基本、最为广泛应用的DNA突变检测技术,PCR可以选择性地将单个DNA或核糖核酸(ribonucleic acid,RNA)分子在几小时内迅速扩增至几百万倍以上,直接检测和分析患者特定基因的序列。从发根、漱口液及少量血痕中得来的极少数细胞均可以直接用于PCR分析,因而可以不必从组织中抽提大量的DNA或RNA样本。
PCR的原理是用一对能分别与靶DNA双链序列配对的人工合成的寡聚核苷酸引物,在耐热DNA聚合酶(Taq polymerase)的作用下,扩增目的DNA片段。重复热变性、引物退火及引物延伸的循环,DNA片段的拷贝数会成指数成倍增长,经过约30多个循环,可达106~107。
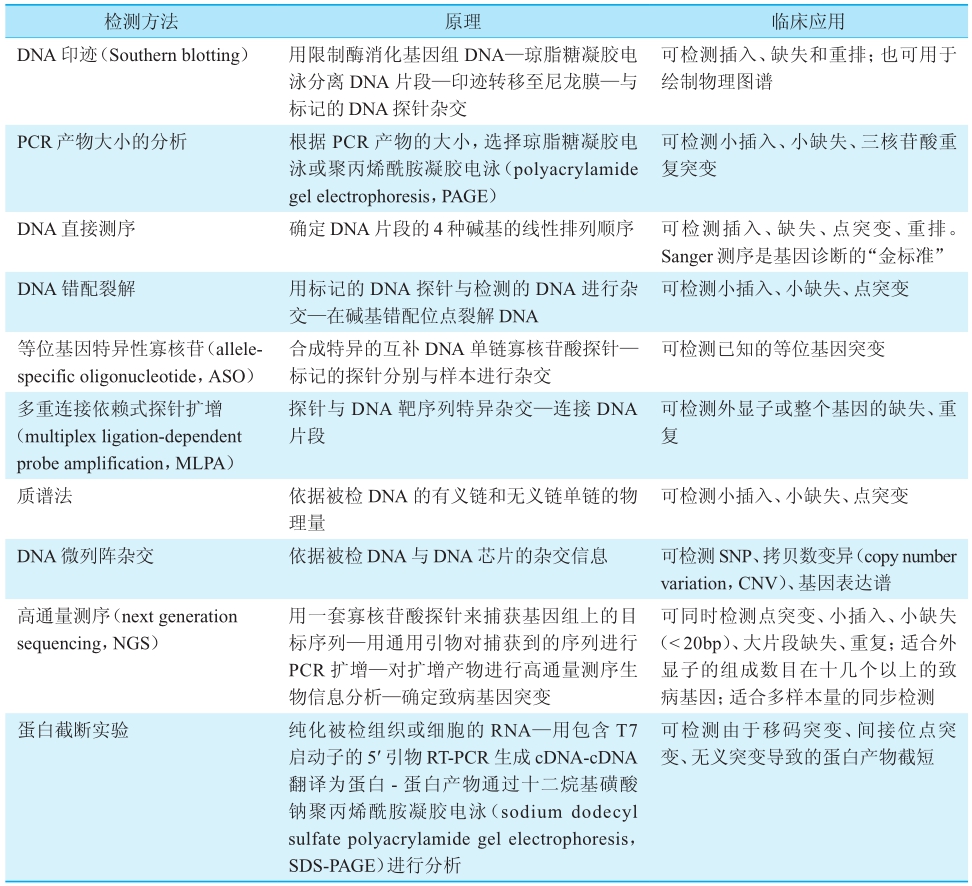
表1 DNA变异检测技术一览[3]
PCR技术有快速、经济、灵敏度高以及对患者核酸检测的样本的要求低等很多优点。但由于PCR的强大扩增能力与检测的敏感性,极微量的污染便可导致假阳性结果。因此,临床实验室要特别重视避免PCR的产物被污染[3,4]。
(二)实时定量聚合酶链反应技术
自从Mullis于1985年发明PCR技术以来,已经衍生出几十种相关的PCR方法,如多重PCR、原位 PCR、实时 -PCR(real time PCR,RT-PCR)和巢式PCR等。定量PCR的目的是以PCR终产物的量推测样本中待测靶分子的绝对起始量和相对起始量,即检测样本中靶基因的拷贝数,该方法对于研究基因的扩增和表达,诊断和判断疾病的预后等都具有非常重要的意义。其中,实时定量 PCR(real time quantitative PCR,qPCR)的应用最为广泛。根据所用荧光技术的不同,qPCR可分为两类:①根据寡核苷酸探针与PCR产物结合后所释放出的荧光进行检测和定量,如TaqMan系统;②通过双链DNA亲和性荧光素与PCR产物结合后所释放出的荧光进行检测和定量,如SYBR Green系统。
在PCR反应的早期,每经过一轮变性、引物退火和延伸合成的周期,DNA分子的数量就会增加一倍。如果对这种相关性进行绘图,在半对数坐标纸上可以得到一个直线图形。达到一定的产量阈值所需的PCR循环数可以用来计算PCR起始模板的数量,通常称为Ct值。达到一定产量所需的循环数越少,起始PCR的模板量就越多[3,4]。
(三)限制性片段长度多态性技术
限制性核酸内切酶是能识别特定的DNA双链序列,并能在识别序列或其邻近处进行DNA双链切割的核酸内切酶,比如EcoRI、BamHI、HaeIII等,都是限制性核酸内切酶。限制性片段长度多态性(restriction fragment length polymorphism,RFLP)是由于DNA序列的变异所引起的限制性内切酶切位点的改变,从而导致酶切片段大小的不同。这种DNA序列的变异由人类基因组中存在的大量单个碱基置换的中立变异以及某些重复序列的重复数目在不同个体中的差异所导致的。这些变异在不同的个体中存在差异,并在人群中表现为遗传多态的现象[3,4]。
(四)脱氧核糖核酸印迹技术
脱氧核糖核酸印迹技术,即DNA印迹(Southern blotting)技术是一种将基因组DNA用限制酶酶切消化后,进行凝胶电泳,将DNA片段按大小进行分离后,然后再用探针进行核酸杂交,以确定目标DNA分子片段位置的一种技术。具体操作如下,首先提取样本基因组DNA,然后限制酶消化,产生约100万个DNA片段,采用琼脂糖凝胶电泳分离DNA片段,小的DNA片段泳动快,而大的DNA片段泳动慢。采用强碱变性的方法,将双链DNA变性而分成两条互补的单链,单链DNA通过印迹作用(blotting)和毛细管作用从凝胶转移到硝酸纤维膜或尼龙膜上。最后,采用经变性后成单链状态的标记DNA探针,与上述通过印迹含有单链DNA分子的膜在液相中进行分子杂交。因为DNA探针只与它的互补DNA链进行复性或退火,所以杂交后的膜洗去那些未结合的探针,然后将膜(膜上已有被杂交上的标有放射性的探针)与X线片压在一起进行曝光,这样X线片上就出现已被杂交的DNA片段的位置,这个位置完全对应于一开始做琼脂糖凝胶电泳时不同样本的DNA 片段的位置 [3,4]。
(五)核糖核酸印迹技术
核糖核酸印迹技术,即RNA印迹(Northern blotting)技术,是用于确定RNA样本中特定基因转录的mRNA分子的大小及含量信息的一种技术。因为不同基因的转录物的长度不同,因此,一种细胞的总RNA或纯化的mRNA可以用琼脂糖凝胶电泳中因分子大小不同而分开,然后同DNA印记技术一样,转移至硝酸纤维素膜或尼龙膜上,将膜与已标有放射性、变性的探针温育杂交,洗膜并与X线片压片曝光,将会出现一个或多个目的转录物的条带[3,4]。
(六)蛋白质印迹技术
蛋白质印迹(Western blotting)技术是用来对特定蛋白质进行定性和半定量的一种方法。本法可用于检测从遗传病患者的细胞中提取的突变蛋白的分子大小及含量的多少。具体操作如下,首先把从细胞中抽提的蛋白质并进行聚丙烯酰胺凝胶电泳(polyacrylamide gel electrophoresis,PAGE),按相对分子质量大小使不同蛋白质分离,然后将它们经印迹转移到一张硝酸纤维素膜、尼龙膜或化学活化膜上,将膜与目的蛋白的抗体一起温育。这种抗原抗体之间的结合可以被标有组织化学、荧光或放射性物质的针对第一抗体的第二抗体所识别。最后,洗膜并与X线片压片曝光,将在目的蛋白质处出现条带,条带的位置和深浅可以用来确定该蛋白分子量的大小和含量的多少 [3,4]。
(七)Sanger测序法
DNA序列测定是诊断已知和未知基因突变最直接、可靠的方法。经典的DNA测序技术称为Sanger测序法(Sanger sequencing),以英国著名科学家、两次诺贝尔奖获得者Fred Sanger命名,是基因诊断的“金标准”。Sanger测序法可用于点突变、小缺失和小插入等的检测。无论是克隆片段还是PCR产物,纯化了的DNA片段都能够进行Sanger测序。Sanger测序法是利用了4个双脱氧核苷酸(ddA、ddC、ddG和ddT),它们的脱氧核糖缺少3′-羟基(通常DNA缺少2′-羟基)。如果加入到正在延伸的DNA链中,双脱氧核苷酸将阻止DNA聚合酶结合到与被测序的模板链互补的下一个碱基,因而阻断DNA的延伸。在Sanger测序中,以被测序的DNA片段作为模板,4种双脱氧核苷酸和正常的4种脱氧核苷酸一并加入反应体系,一段短寡核苷酸引物按照碱基互补配对的原则,结合到DNA模板上,DNA聚合酶会选择任一正常核苷酸或双脱氧核苷酸加入而继续延伸合成链。因为如果加入双脱氧核苷酸将而终止DNA链的合成。因此,PCR结束后,理论上反应体系中应该存在不同长度片段(只相差一个碱基)的DNA产物,用电泳的方法分离DNA产物,因为每一种双脱氧核苷酸标记有可发出不同荧光的荧光染料,因此,根据不同的荧光标记,就可读出DNA的碱基序列。将该序列与网上数据库中的正常序列进行比对,可以明确是否存在目的DNA序列的点突变、小缺失和小插入等[3,4]。
(八)高通量测序技术
高通量测序又称二代测序(next generation sequencing,NGS)或大规模平行测序技术,对应于以Sanger测序法为代表的第一代测序技术而得名。在二代测序中,三种主流测序技术分别为依次出现的Roche/454焦磷酸测序(2005年)、Illumina/Solexa聚合酶合成测序(2006年)和ABI/SOLiD连接酶测序(2007年)。高通量测序可检测全基因组存在的点突变、小插入或者缺少等。与Sanger测序相比,二代测序技术最突出的特征是单次运行能够产出巨大的序列数据量。
高通量测序技术一般由模板准备、测序和成像、序列组装和比对等部分组成。三种高通量测序技术的原理各不相同,其数据量、数据质量和单次运行的成本也有差异。相对于Sanger测序技术,高通量测序技术的出现,使得获得核酸序列数据的单碱基测序费用急剧下降,同时也给基因组学研究带来了新方法和新方案。目前,高通量测序技术已广泛应用于动植物全基因组测序、基因组重测序、外显子组测序、转录组测序、小RNAs测序和表观基因组测序等方面,并在孟德尔疾病和复杂疾病的研究以及疾病的基因诊断和靶向药物应用中发挥重要作用[5~10]。
(九)多重连接依赖式探针扩增技术
多重连接依赖式探针扩增(multiplex ligationdependent probe amplification,MLPA)技术是一种高通量、针对待测DNA靶序列进行定性和半定量分析的方法。该技术具有高效、特异、在一次反应管中可同时检测多个不同的核苷酸序列拷贝数变化的优点,可用于检测DNA的大片段缺失或者重复。MLPA的基本实验流程和原理包括DNA变性、探针与DNA靶序列杂交、连接、PCR扩增,产物通过毛细管电泳分离后,最后采用软件分析获得DNA靶序列重复或者缺少的结论。
每对MLPA探针包括两段寡核苷酸序列,每条探针包括一段引物序列和一段特异性序列。通过与靶序列的杂交,并使用连接酶把两部分探针连接成一条核苷酸单链,再通过通用引物进行扩增。由于设计的每对探针所扩增的产物长度不一,毛细管电泳可将PCR产物的不同片段进行分离,最后应用Genemarker等软件对结果进行分析。如果检测的靶序列发生点突变、甲基化、缺失、重复等变异,则相应探针的扩增峰便会发生缺失、降低或升高。因此,根据扩增产物的改变,可判定靶序列是否存在拷贝数目的异常、点突变和甲基化等现象 [3,4]。
二、DNA突变的预筛查技术
对于未知基因突变的筛查,有以下几种技术:①基于突变型和野生型片段单链在凝胶电泳上迁移率的不同或指纹图谱差异的检测,最典型的是单链构象多态性(single-strand conformational polymorphism,SSCP)、双脱氧指纹法(dideoxy fingerprinting,ddF)和限制性内切酶指纹法(restriction endonuclease fingerprinting,REF)等;②基于突变型和野生型片段生成的杂合双链体与纯合双链体在PAGE上迁移率不同的检测,包括变性梯度凝胶电泳(denaturing gradient gel electrophoresis,DGGE)、温度梯度凝胶电泳(temperature gradient gel electrophoresis,TGGE)、构象敏感凝胶电泳(conformation-sensitive gel electrophoresis,CSGE)和异源双链分析(heteroduplexes analysis,HA)等;③基于杂合双链体和纯合双链体在高效液相色谱中滞留时间的差异的筛查方法,即变性高效液相色谱(denaturing high performance liquid chromatographty,DHPLC)分析;④基于杂合双链体中错配碱基的化学断裂法(chemical cleavage of mismatch,CCM),如用核糖核酸酶(ribonuclease,RNase),DNA修复酶或化学试剂(四氧化锇、羟胺)的检测。上述检测方法对于未知基因突变的筛查在灵敏度、适用范围和检测突变类型上存在差异,各有优势和不足[1,2]。
三、间接检测DNA突变的技术
一般将基因诊断的方法分为直接法和间接法两大类。直接法如上所述,主要是直接对致病基因进行诊断和分析,检查其是否存在结构异常。而间接分析法主要是用基因旁边或内部的遗传多态标记在家系中作连锁分析而进行基因诊断,主要是用多态性位点来跟踪致病基因的传递。间接检测DNA突变的技术一般在下述情况下采用:①致病基因还未得到完全克隆;②患者的家系资料较为完整;③已知与致病基因紧密连锁的DNA多态性位点。
(一)短串联重复序列连锁分析技术
短串联重复序列(short tandem repeat,STR)是遍布于人基因组中的高度重复序列,重复单位一般为2~6bp,又称为微卫星DNA(microsatellite DNA)。在人群中因重复次数不同而存在遗传多态性,它的杂合度和所含信息都比较高,因此,可应用该信息对未知致病基因的遗传病进行诊断,也可以作为直接检测DNA突变的验证手段之一。在采用STR作连锁分析进行基因诊断时,发生错误诊断的概率大小与突变基因与多态遗传标记间的重组有关 [3,4]。
(二)DNA指纹
长度10~100bp的DNA高度重复序列(重复次数通常为几百至几千次)称为小卫星DNA(minisatellite DNA)。由于小卫星可变的串联重复次数造成许多等位基因,故又称为可变数目串联重复(variable number of tandem repeat,VNTR)。这种信息量最大的多态性标记可涉及几十个或以上的等位基因。因而,如果两个个体之间无亲缘关系,她们的等位基因就不一样。无论是STR还是VNTR,大部分可能对健康无影响,但有些VNTR可引发相关疾病的发生[3,4]。
参考文献
1.陈竺. 医学遗传学[M].北京:人民卫生出版社,2005.
2.邬玲仟,张学.医学遗传学[M].北京:人民卫生出版社,2016.
3.VAN OMMEN GJB,BAKKER E,DEN DUNNE J T. The human genome project and the future of diagnostics,treatment,and prevention[J]. Lancet,1999,354(Suppl 1):5-10.
4.GOODWIN S,MCPHERSON J D,MCCOMBIE W R. Coming of age:ten years of next-generation sequencing technologies[J]. Nat Rev Genet,2016,17(6):333-351.
5.SHENDURE J,BALASUBRAMANIAN S,CHURCH G M,et al. DNA sequencing at 40:past,present and future[J]. Nature,2017,550(7676):345-353.
6.BIESECKER L G,GREEN R C. Diagnostic clinical genome and exome sequencing[J]. N Engl J Med,2014,370(25):2418-2425
7.COLLINS F S,VARMUS H. A new initiative on precision medicine[J]. N Engl J Med,2015,372(9):793-795.
8.TUCKER T,MARRA M,FRIEDMAN JM. Massively parallel sequencing:the next big thing in genetic medicine[J].Am J Hum Genet,2009,85(2):142-154.
9.HEATHER J M,CHAIN B. The sequence of sequencers:The history of sequencing DNA[J]. Genomics,2016,107(1):1-8.
10.BEHJATI S,TARPEY P S. What is next generation sequencing?[J]. Arch Dis Child EducPract Ed,2013,98(6):236-238.
知识来源
来源:人卫知识数字服务体系
作者:龚凤英,中国医学科学院北京协和医院内分泌科
专家简介
研究员:龚凤英
国家卫生健康委内分泌重点实验室(北京协和医院)学术委员会实验室专职人员
- 评价此内容

 3我要打分
3我要打分



 会员登录
会员登录
近期推荐
- 顾洪斌委员:糖尿病足的临床诊…
- 文富强副主委:糖皮质激素临床…
- 李东晓教授:肥胖的预防和治疗
- 蒋铁建、雷闽湘:详解卡尔曼综…
- 朱惠娟、刘小海:一文详述垂体…
- 陈慧玲、雷闽湘:下丘脑-垂体…
- 禹松林:液相色谱串联质谱技术…
- 龚凤英:基因突变的几种常见检…
- 卢琳教授:内分泌激素检测结果…
- 张爱珍教授:肥胖症的治疗

热门关键词
最新会议
- 2013循证医学和实效研究方法学研讨会
- 欧洲心脏病学会年会
- 世界帕金森病和相关疾病2013年会议
- 英国介入放射学学会2013年第25届年会
- 美国血液学会2013年年会
- 美国癫痫学会2013年第67届年会
- 肥胖学会 2013年年会
- 2013年第9届欧洲抗体会议
- 国际精神病学协会 2013年会议
- 妇科肿瘤2013年第18届大会
- 国际创伤压力研究学会2013年第29届…
- 2013年第4届亚太地区骨质疏松症会议
- 皮肤病协会国际2013年会议
- 世界糖尿病2013年大会
- 2013年国际成瘾性药年会
- 彭晓霞---诊断试验的Meta分析
- 武姗姗---累积Meta分析和TSA分析
- 孙凤---Network Meta分析
- 杨智荣---Cochrane综述实战经验分享
- 杨祖耀---疾病频率资料的Meta分析
友情链接
合作伙伴
Copyright g-medon.com All Rights Reserved 环球医学资讯 未经授权请勿转载!
网络实名:环球医学:京ICP备08004413号-2
关于我们|
我们的服务|版权及责任声明|联系我们
互联网药品信息服务资格证书(京)-经营性-2017-0027
互联网医疗保健信息服务复核同意书 京卫计网审[2015]第0344号
