朱惠娟、包新杰:一文详解垂体生长激素细胞腺瘤
垂体生长激素细胞腺瘤是指来源于分泌生长激素(growth hormone,GH)腺垂体细胞的腺瘤,是功能性垂体腺瘤中较为常见的一种类型。发生在青少年骨骺闭合之前的GH过度分泌导致巨人症,发生在骨骺闭合后的成年人则主要表现为肢端肥大症。1886年法国神经学家Pierre Marie首次使用“Acromegaly(肢端肥大症,简称肢大)”一词描述具有典型肢端肥大表现的患者。垂体生长激素腺瘤患者除了有生长激素和类胰岛素样生长因子1(insulin-like growth factor 1,IGF-1)高分泌的特征性临床表现外,还常伴有糖代谢异常、高血压、骨关节疼痛等相关的并发症,控制不佳的垂体GH腺瘤患者发生心脑血管疾病的风险显著增加。
一、流行病学
我国垂体生长激素细胞腺瘤患者的临床流行病学数据不详。各国的患病率和发病率的报道也不尽相同。
2000年前,全球报道的年患病率稳定低于7/100 000。但近5年,冰岛和马耳他共和国的报告年患病率超过13/100 000。但与此同时,瑞典、西班牙和丹麦等国基于国家注册研究数据显示年患病率降低为3.6~3.9/100 000[1-8]。在1980—1990期间,全球每年的发病率约0.4/100 000~1.28/100 000,近期报告约为1.1/100 000[1-5]。随着越来越多的内分泌和其他专科医生对肢端肥大症认识的深入,特别是2000年后对肢端肥大症诊断方法的推广普及以及社会媒体和网络的宣传,该疾病发现率逐渐增加。在临床中特定人群筛查肢端肥大症也十分重要,如:研究发现,在睡眠呼吸暂停综合征(sleep apnea,OSAS)患者筛查中,肢端肥大症的患病率约为2.5/1 000[9]。肢端肥大症患者的平均诊断年龄40~50岁。诊断延迟十分常见。从出现症状到临床诊断的中位时间约为5年,甚至有长达25年的病例报告。虽然有病例报告男性发病率略高于女性,但大样本研究未发现不同性别的患病率有显著差异。与肢端肥大症相比,垂体性巨人症的病例更为罕见,北京协和医院在1990—2017年间共诊治垂体性巨人症56例。
二、病理生理
GH是由腺垂体的生长激素细胞合成并分泌的,受下丘脑分泌的生长激素释放激素(growth hormone releasing hormone,GHRH)和生长抑素的共同调节。GHRH从神经元轴突释放后进入垂体门脉血管到达腺垂体,作用于生长激素细胞的GHRH受体,促进GH的合成和分泌。生长激素基因位于第17对染色体的长臂,编码含有191氨基酸的GH。生理状态下,GH呈脉冲式分泌,夜间入睡后分泌的峰值和频率显著高于日间,不同年龄段GH脉冲频率和幅度也不同,青春发育期的青少年生长激素的脉冲频率显著多于成人。同时血清生长激素浓度也受运动、应激及葡萄糖等代谢物质影响。分泌入血的GH作用在肝细胞表面的GH受体刺激肝细胞合成和分泌IGF-1。GH和IGF-1均可通过负反馈调节GH的分泌。在青少年骨骺未闭合之前,GH可通过IGF-1介导促进骨骼生长。此外,GH还是调节糖代谢、脂肪和蛋白质代谢的主要激素,体内过量分泌的GH可减少葡萄糖的摄取,并促进蛋白质的合成和脂肪的分解。
机体GH过度分泌多是因垂体GH细胞腺瘤或鞍区异位分泌GH的腺瘤过度分泌所致,亦可能是下丘脑或者异位分泌过多GHRH间接刺激垂体分泌过多的GH导致,其中,垂体GH细胞腺瘤约占95%以上。随着基因检测技术的发展,越来越多的单基因突变被发现与垂体GH细胞腺瘤相关,特别是儿童或青少年起病的巨人症患者。也有罕见病例是因为胰腺和肺类癌异位分泌GHRH或GH导致肢端肥大症表现。目前发现的与垂体GH细胞腺瘤相关的基因有:
1.芳香烃受体相互作用蛋白基因
在正常的腺垂体组织中,芳香烃受体相互作用蛋白(aryl hydrocarbon receptor-interacting protein,AIP)基因主要在分泌GH和泌乳素(prolactin,PRL)的垂体细胞上表达,与该基因突变相关的疾病为家族孤立性垂体腺瘤(familial isolated pituitary adenoma,FIPA),定义为家族中有2个或者2个以上成员患有垂体腺瘤,不伴有其他器官的受累。家族单纯性垂体腺瘤(familial isolated pituitary adenoma,FIPA)占垂体肿瘤的2%~3%,而AIP基因突变是其最常见的致病基因,AIP突变导致的垂体腺瘤常起病较早,肿瘤的体积更大[10,11]。
2.G蛋白偶联受体101基因(GPR101基因)
2015年Trivellin,G.等首次在14例巨人症患者中发现G蛋白偶联受体101基因(G-protein coupled receptor 101,GPR101)突变,研究发现巨人症与Xq26.3染色体片段上GPR101基因的微重复有关,但GPR101基因突变引起GH分泌增加的机制不详。目前将这种X染色体相关的基因突变引起的巨人症命名为X-染色体相关的肢端肥大性巨人症(X-linked acrogigantism,X-LAG)[12]。
3.GNAS基因
GNAS基因位于20号染色体长臂,编码G蛋白偶联受体α亚单位。体细胞GNAS基因突变是McCune-Albright综合征(McCune-Albright Syndrome,MAS)的发病原因,G蛋白偶联受体的自主活化导致MAS患者出现内分泌腺体功能亢进的临床表现。约20%~30%的MAS患者垂体受累,包括垂体生长激素细胞增生、垂体生长激素腺瘤、泌乳素瘤、ACTH腺瘤等。研究发现约15%的垂体生长激素细胞腺瘤患者的肿瘤组织中可以检测到GNAS基因的突变[13,14]。
4.多发性内分泌腺瘤病1基因(MEN1基因)
多发性内分泌腺瘤病1基因(multiple endocrine neoplasia 1,MEN1)位于 11q13上,编码核蛋白menin。MEN1基因突变导致常染色体显性遗传病多发性内分泌腺瘤病1型(multiple endocrine neoplasia1,MEN1)。约有90%的家族性MEN1可检测到该基因的突变,该病的临床表现主要跟受累的内分泌腺体有关,临床表现为甲状旁腺增生或腺瘤导致的功能亢进,胰腺神经内分泌肿瘤如胃泌素瘤、胰岛β细胞瘤或胰高血糖素瘤。垂体腺瘤约占10%~60%,其中,约80%的垂体腺瘤为垂体大腺瘤,可分泌催乳素、生长激素、促肾上腺皮质激素,生长激素瘤占7%左右,表现为肢端肥大症、巨人症等。研究也发现此类垂体腺瘤更具有侵袭性,无论是对于临床诊断还是治疗也更有挑战[15,16]。北京协和医院回顾性总结54例合并垂体瘤的MEN1患者,其中无功能瘤最常见,约占48.1%,其中9.3%(5/54)为生长激素腺瘤[17]。
5.细胞周期依赖性激酶1B基因(CDKN1B基因)
细胞周期依赖性激酶1B基因(eyclin-dependent kinase inhibitor 1B,CDKN1B)主要编码细胞周期依赖性激酶(CDK)抑制蛋白p27,这种基因突变导致的疾病的临床症状与MEN1相似,但检测不到MEN1突变,又被称作MEN4,大约占临床表现为MEN1症状的患者的1%~2%。其引起的垂体腺瘤中最常见的是垂体生长激素细胞腺瘤。
6.蛋白激酶A的调节亚单位1α基因(cAMP-dependent protein kinase type I-alpha regulatory subunit,PRKAR1A基因)
蛋白激酶A的调节亚单位1α基因(cAMP-dependent protein kinase type I-alpha regulatory subunit,PRKAR1A)位于17q22-24,编码蛋白激酶A的调解亚单位,作为一种抑癌基因,突变可导致多种肿瘤的发生。约44%的Carney综合征的发生与该基因突变相关,Carney综合征是一种常染色体显性遗传的多发性肿瘤综合征,主要表现包括皮肤点状色素沉着、心房黏液瘤和内分泌腺体肿瘤,包括原发性色素结节性肾上腺皮质病(primary pigmented nodular adrenocortical disease,PPNAD)、垂体生长激素细胞腺瘤、睾丸大细胞钙化性支持细胞瘤、甲状腺结节(癌)和卵巢囊肿等[18,19]。
7.琥珀酸脱氢酶D基因(SDHD基因)
琥珀酸脱氢酶D基因(succinate dehydrogenase D,SDHD)位于11q23,研究发现8.9%的副神经节瘤患者存在该基因突变,其中有患者合并垂体腺瘤,其中最常见的类型为泌乳素细胞腺瘤,其次是垂体生长激素细胞腺瘤。当患者诊断为垂体生长激素细胞腺瘤,合并副神经节瘤的证据或家族史时,可进一步筛查SDHD基因[20]。
三、临床表现
垂体GH细胞腺瘤临床表现包括肿瘤压迫周围组织或正常腺垂体表现、高分泌GH和IGF-1导致的症状,少数患者可能出现垂体卒中的临床表现。
1.肿瘤压迫周围组织和正常腺垂体的相关临床症状
常见于垂体GH细胞大腺瘤或巨腺瘤,肿瘤压迫鞍膈导致眼后、额颞部的头痛。少数患者肿瘤向鞍上发展,压迫第三脑室和室间孔,可引起梗阻性颅压升高,出现全头疼痛。微腺瘤可通过“窃血”使视交叉中部血供相对减少,大腺瘤可直接压迫上抬视交叉的神经纤维及相关血管导致视功能障碍,典型的临床表现为视力下降、颞侧视野缺损致半盲甚至全盲。若肿瘤向两侧侵及海绵窦,部分患者还可有第Ⅲ、Ⅳ、Ⅵ对脑神经的受累,可表现为眼球运动障碍、眼睑下垂、瞳孔散大或对光反应迟钝等。垂体GH细胞大腺瘤的患者可能会压迫正常的腺垂体导致部分腺垂体的功能低下,出现相关临床表现,如:月经紊乱和闭经、性功能减退、怕冷、便秘、乏力、食欲缺乏等。
2.GH高分泌相关的临床表现
GH过量分泌引起的临床表现比较隐匿,常在病程持续较长时间才被察觉,多数患者是因其并发症而被诊断,如睡眠呼吸暂停(sleep apnea,OSAS)、糖尿病、高血压等。
(1)骨骼和软组织改变
循环中,高GH和IGF-1水平会导致骨骼的改变。在骨骺闭合前,外周循环中IGF-1持续作用于软骨细胞促进新骨形成,导致生长持续加速。表现为身材高大,显著高于同族同龄正常儿童或青少年身高97百分位以上。而成人则表现为扁骨和软组织的过度生长,包括眉弓突出、前额变宽、下颌突出、反咬合、齿列稀疏、鼻翼增大等面容改变及显著的手足增大。咽喉软组织增生患者可有声音低沉。骨关节病在垂体GH细胞腺瘤患者中非常常见,肘关节、髋关节、肩关节、腰骶关节均可受累,可表现为关节肿胀、软骨变厚,超过一半的患者有活动受限,50%患者因软组织增生和压迫出现腕管综合征。
肢端肥大症患者过量分泌的GH/IGF1通过增加骨吸收和影响骨微结构导致椎体骨折(vertebral factures,VFs)风险显著增加。肢端肥大症患者因维生素D结合蛋白水平的显著增加导致游离维生素D水平显著降低。而GH过量可使甲状旁腺激素(parathyroid hormone,PTH)脉冲分泌的时相延长,脉冲分泌量增加。GH刺激小肠钙磷的吸收。因此,活动期肢大患者常表现出轻度的钙磷代谢异常,包括轻度的高磷血症和血钙水平偏高[21]。
(2)皮肤改变
肢端肥大症患者早期的皮肤改变主要表现为皮肤多汗和油脂分泌旺盛,皮肤逐渐变厚、粗糙,额部出现皱褶、鼻翼肥大、鼻唇沟加深。
(3)心血管并发症
心血管疾病是肢端肥大症患者最主要的死亡原因。主要的心血管异常包括高血压、心脏肥大及左心室功能不全、冠状动脉硬化性心脏病及心律不齐等。GH增加远端肾小管和集合管对钠离子的重吸收,血容量的增加,肢端肥大症患者的高血压患病率显著高于正常人群,超过半数的肢大患者发生临界高血压或高血压。心脏肥大是肢大患者常见的表现,北京协和医院经超声心动图测定的76例肢大患者经体表面积校正后的心肌重量达(339 ± 86)g/m2,显著高于正常人的(121 ± 12)g/m2。除了心肌肥厚,病理检查发现心肌出现局限性间质纤维化、单核细胞浸润或典型心肌炎的表现,提示肢大患者的心脏肥大并非单纯的GH的促蛋白合成作用所致,还有其他的机制参与肢大心肌病的发生中来,因此,病程较长的肢大患者易发生心功能不全、心律失常。GH对葡萄糖代谢、脂质代谢有影响,肢大患者更容易罹患冠状动脉粥样硬化型心脏病。约3%的肢端肥大症患者出现严重的心脏并发症如:充血性心衰等,甚至危及生命[22]。
(4)呼吸系统并发症
GH和IGF-1可以直接刺激呼吸道黏膜增生、颈部软组织的增生,舌体肥大、颌骨突入等导致呼吸道结构发生改变,导致肢大患者常常合并呼吸系统功能障碍,呼吸系统并发症的严重程度和病程及生长激素的水平相关。文献报道,60%~80%的肢大患者合并阻塞性呼吸睡眠暂停的相关临床表现,其中男性更为常见。肢大患者在麻醉、术后拔管和急性呼吸道感染的时候易出现呼吸道阻塞[23]。但北京协和医院关于肢大患者呼吸多导睡眠仪监测结果发现,42.6%的患者合并阻塞性呼吸暂停低通气。
(5)神经肌肉系统改变
肢大患者较常出现双手麻木疼痛、肌力下降等症状,神经检查可发现正中神经运动和感觉传导异常,软组织增生、腕关节软骨和肌腱压迫正中神经引起的腕管综合征。长期病情活动的肢大患者可出现活动耐力的下降,肌酶水平正常,肌电图可有肌病的表现,有研究发现肌肉活检显示Ⅱ型肌纤维萎缩,Ⅰ型肌纤维增生。有效控制GH水平后肌力可逐渐改善。
(6)内分泌并发症
GH与糖代谢关系密切,既有类胰岛素作用,又有拮抗胰岛素作用。持续GH水平增高导致糖耐量异常如糖耐量受损或糖尿病,甚至部分患者因显著高血糖、糖尿病酮症酸中毒起病后诊断发现为垂体GH细胞腺瘤。研究发现20%~56%的肢端肥大症患者合并糖尿病,16%~46%合并糖耐量低减[24]。当肢端肥大症患者有效治疗后,患者糖代谢状态也会有不同程度的改善。
(7)肿瘤
既往研究表明,肢端肥大症与部分良性和恶性的肿瘤的发生有关。45%的肢端肥大症患者患有结肠息肉,其中,24%为结肠腺瘤。因此,推荐肢端肥大症患者常规进行结肠息肉和结肠癌的筛查[25]。肢端肥大症患者发生甲状腺结节和甲状腺乳头状癌的发生风险也显著增加。
(8)混合型垂体腺瘤
垂体GH、PRL和促甲状腺激素(thyroid stimulating hormone,TSH)可以同时出现高分泌状态,既可能是同一细胞表达不同激素的分泌颗粒,也可能是腺瘤内有分泌不同激素的细胞共同存在。合并高PRL血症患者可表现为泌乳、闭经、性功能减退和不育等。合并TSH不适当分泌的患者可有怕热、多汗、易饥、心悸等甲亢的表现。
(9)垂体卒中
垂体GH腺瘤起病隐匿,往往诊断时就为大腺瘤,部分患者可有垂体腺瘤自发的出血、坏死,即垂体卒中。临床主要表现为剧烈的头痛、恶心、呕吐、视野缺损等表现。
临床上需要筛查垂体GH腺瘤的患者包括:新发糖尿病患者;多发关节疼痛的患者;新发或难以控制的高血压患者;发现心室肥大或收缩、舒张功能障碍等心脏疾病的患者;无明显诱因出现乏力、头疼、腕管综合征、睡眠呼吸暂停综合征、多汗、视力下降、结肠息肉和进展性下颌突出的患者[26]。
四、病理诊断
根据2017年WHO最新垂体肿瘤病理分型指南,过度分泌GH的垂体肿瘤命名为垂体GH细胞腺瘤,其来源于Pit-1腺垂体细胞系,主要分为以下几种亚型:致密颗粒型GH细胞腺瘤(DGSAs)、稀疏颗粒型GH细胞腺瘤(SGSAs)、PRL-GH细胞腺瘤、混合性PRL-GH细胞腺瘤,前两者的区别在于低分子量细胞角蛋白(LMWK)的分布,DGSAs中LMWK在核周或者弥漫分布,而其在SGSAs中呈点状分布。DGSAs和SGSAs在影像学特征和治疗反应上也有区别,SGSAs在T2加权相上为信号更高,多为侵袭性生长,且其对生长抑素类似物治疗不敏感;相反,DGSAs在T2加权相上通常信号较低,更易发现GNAS突变,对生长抑素类似物治疗敏感。后两者分别代表在相同细胞和不同细胞可观察到GH和PRL免疫组化染色的阳性。新的WHO病理分型提出多激素细胞腺瘤的概念,指表达一种以上腺垂体激素的腺垂体细胞增生,来源于一种或多种腺垂体细胞系,并指出,稀疏颗粒型GH细胞腺瘤、多激素型Pit-1阳性垂体腺瘤均属于高危垂体腺瘤。Ki67等免疫组化染色有助于了解垂体腺瘤细胞的增殖能力[27]。
五、定性诊断
肢端肥大症的定性诊断主要包括临床表现和生化检测。具有典型肢端肥大症临床表现的患者应接受生长激素和IGF-1的检查明确诊断,临床上也要关注临床表现不典型患者的筛查,以期早诊断,提高治愈率,如:糖尿病、高血压、OSAS、关节炎、腕管综合征和不明原因多汗、乏力的患者。生化检测主要包括如下内容:
1.血清IGF-1水平测定
血清中IGF-1主要与IGF-1结合蛋白结合,其半衰期长,血浓度波动小,不仅可用于诊断肢端肥大症,也是反映垂体GH腺瘤活动性的重要血清标志物,若其高于同性别同年龄血清IGF-1水平正常上限,则高度提示有高GH血症的可能。但需要注意的是IGF-1水平也受到营养状态、肝功能、测定方法的影响,青春期和妊娠期正常人的IGF-1水平会有一定程度的增加,而血糖控制不佳的糖尿病患者和肾衰患者的IGF-1水平会有所下降,因此,需综合分析结果。
2.口服葡萄糖GH抑制试验
空腹GH水平个体差异较大,而且GH作为应激激素受到运动、情绪等影响.因此,口服葡萄糖GH抑制试验可作为肢端肥大症GH高分泌状态的诊断试验。具体试验方法:口服75 g葡萄糖,分别在0、30、60、90及120分钟取血测定血糖及GH水平。如果GH谷值水平< 1ng/ml,判断为GH水平被正常抑制。若谷值≥1ng/ml则视为不能被抑制,提示体内有自主GH分泌,可进一步接受肢端肥大症的定位诊断。虽然化学发光免疫测定方法显著增加了GH测定的灵敏度,也有学者提出0.4ng/ml作为诊断的界值,但目前国内外的指南仍以GH谷值< 1ng/ml作为正常抑制标准[26,28]。
需要和肢端肥大症鉴别诊断的情况包括:部分患者有较为典型的临床表现,但GH/IGF-1等生化指标正常,影像学可能阴性甚至表现为空泡蝶鞍,这种情况要注意问诊患者是否有剧烈头痛的病史,以除外肿瘤卒中。肢大患者还需和肥厚型骨关节病(原发性肥厚性骨关节病)相鉴别,后者多因基因缺陷(如HPGD缺乏,SLCO2A1缺乏)导致前列腺素E2显著升高,患者可以出现类似肢大的表现,但手足表现不典型,以杵状指和关节肿胀为主,GH/IGF1水平正常;部分严重胰岛素抵抗的患者也有类肢端肥大症临床表现,但GH/IGF-1水平正常,垂体影像学阴性。
六、定位诊断
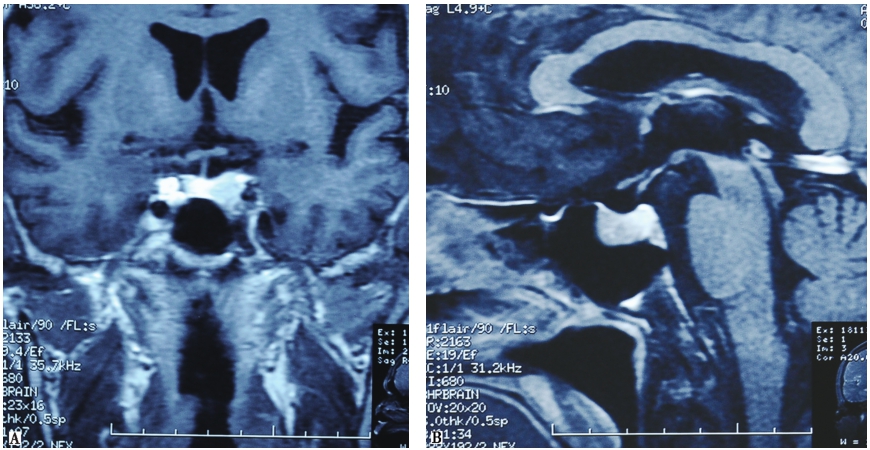
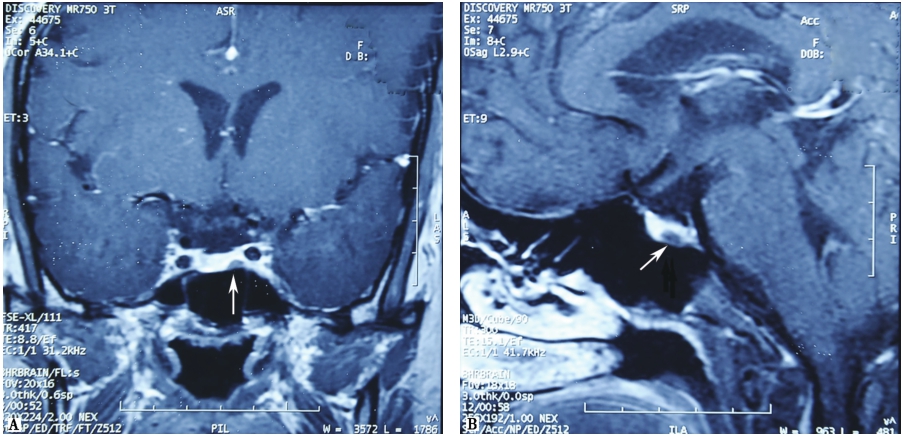
当实验室检查提示体内GH分泌过多,则需要进一步完善影像学相关的检查,以辅助疾病定位诊断。为明确垂体GH腺瘤与毗邻组织的关系,肿瘤大小、是否呈侵袭性生长,肿瘤和海绵窦的关系以及视交叉是否受累,并进行头部影像学检查等,对术前和术中判断非常帮助。磁共振的分辨力优于CT(图1)。进行鞍区的动态及高分辨的薄层MRI增强扫描检查可增加垂体微小GH腺瘤的检出率。
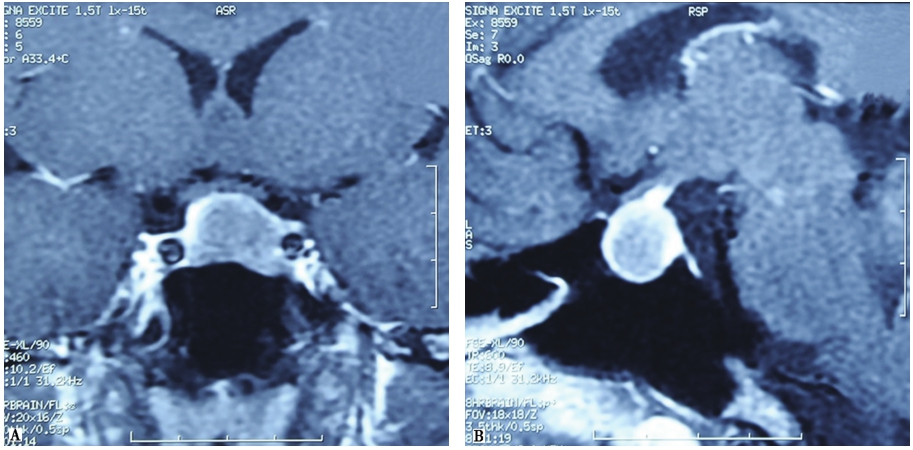
图1 垂体GH大腺瘤MRI增强扫描
鞍内可见约2.0cm×1.3cm×1.5cm类圆形低信号影,见视交叉受压,正常垂体受压变薄,左侧颈内动脉海绵窦段被病变部分包绕,造影剂增强后轻度强化。A.冠状位;B.矢状位
七、治疗
(一)治疗目标
垂体GH腺瘤的治疗目标有以下5方面[26]:①血清GH水平OGTT:GH谷值< 1.0μg/L;②IGF-1降至与年龄和性别相匹配的正常范围;③尽可能消除或者缩小垂体GH腺瘤,防止肿瘤复发;④消除或减轻患者临床症状及并发症;⑤尽量保留腺垂体的内分泌功能,对有垂体功能减退的患者行替代治疗。
(二)治疗方法
1.手术治疗
垂体GH腺瘤患者首选手术治疗。对于手术有可能治愈、以局灶生长方式的大腺瘤以及垂体微腺瘤患者,因为手术治疗可长期且有效控制肿瘤,并使相关的生化指标达到正常水平,故其一线治疗方案是手术。
在垂体GH腺瘤患者中,与传统开颅手术相比,经鼻蝶窦入路手术切除术更安全有效,死亡率低且并发症少。对于伴有急性且严重的肿瘤压迫症状及垂体功能减退的患者,建议及早手术治疗。对伴有肿瘤压迫相关症状但手术治愈困难的垂体GH大腺瘤患者,可先行部分切除术,以提高下一步药物治疗或者放疗的疗效。成功切除肿瘤不仅可以缓解肿块引起的压迫症状,还可以有效降低GH水平。研究发现,患者术前的IGF-1和GH水平,肿瘤侵袭性、大小、质地是影响手术疗效的重要因素。对于术前GH和IGF-l水平仅略高于正常且肿瘤未侵袭海绵窦的微腺瘤患者,手术治愈率可以达到80%。但对术前GH > 200μg/L或肿瘤已侵袭海绵窦的患者,手术治愈的可能性小。部分患者可在术前行SSA治疗,以为手术创造条件,提高手术效果。
2.药物治疗
临床用于肢端肥大症治疗的药物包括生长抑素受体配基(生长抑素类似物)如奥曲肽、兰瑞肽和帕瑞肽、多巴胺受体激动剂和GH受体拮抗剂培维索孟。
(1)长效生长抑素类似物
药物治疗主要用于不适合立即接受手术的患者,包括:全身状况较差难以承受手术风险的患者;因肢大的气道问题麻醉风险较高的患者;有严重的肢大全身表现如:心肌病、重度高血压和未能控制的糖尿病等的患者,也可首选药物治疗。长效生长抑素类似物能够使GH/IGF-1下降,显著改善肢端肥大症的临床并发症,特别是对软组织的作用显著,改善患者的心肺功能,同时可以使超过半数的患者GH和IGF-1水平正常,肿瘤缩小,提高手术的安全性。其次药物可以用于术后残余肿瘤的治疗,以及残余肿瘤放射治疗后,等待放疗充分发挥作用时SSAa进行过渡期的治疗。SSA的不良反应:SSA的不良反应主要为注射部位反应和胃肠道症状。5%~15%的患者有胃肠道症状,腹泻、腹痛、腹胀、脂肪泻、恶心和呕吐。长期使用SSAa可以使胆囊淤泥或胆结石发病率增加,通常没有症状,大多数不需要手术干预,可定期超声检测。少见的不良反应还包括脱发、心动过缓和便秘。
(2)多巴胺受体激动剂:
多巴胺受体激动剂可以通过下丘脑的多巴胺受体而抑制GH的释放。常用的多巴胺受体激动剂包括麦角衍生物溴隐亭和卡麦角林,GH水平轻中度升高的患者使用这类药物后,有10%~20%的患者GH和IGF-1水平显著降低。多巴胺受体激动剂的不良反应包括胃肠道不适、直立性低血压、头痛、鼻塞和便秘等。目前,国内仅有第一代多巴胺受体激动剂溴隐亭,该药适合用于GH水平轻度升高而由于其他原因未能使用SSA的患者。
(3)GH受体拮抗剂(培维索孟):
此类药物通过直接抑制GH受体降低IGF-1的生成发挥直接改善临床症状的作用,但并未直接抑制肿瘤的生长,常需要和生长抑素类药物联合使用。
3.放射和放射外科治疗
因放疗后患者的GH值一般下降比较缓慢,并且可能有垂体功能低下等并发症,故放疗不作为治疗的首选方式。而放疗最常用于术后肿瘤残留和复发的辅助方案。对于术后仍存有GH高分泌或者不能进行手术的患者可选择放疗。传统的分次放疗过去主要用于控制肿瘤生长和达到生化缓解,通常起效时间需要6个月至2年,部分需5~15年才能完全发挥作用。最近研究发现,大剂量定向放疗(包括质子束治疗以及立体定向放射外科治疗)对垂体残余肿瘤的疗效显示,与传统的放疗比较,立体定向放射外科治疗(如伽玛刀)及立体定向放射治疗后患者的病情缓解更快[29]。放疗最常见的并发症是腺垂体功能受损(30%左右),传统放疗的发生率较高,其发生后通常需要行激素替代治疗。
应根据垂体GH腺瘤患者及所在医院垂体腺瘤治疗方面的综合情况制定个体化的治疗策略。所有治疗策略最终目的都应是控制患者的GH水平。在缓解垂体GH腺瘤的压迫效应以及控制生化指标的同时,要全面综合评估每位患者的治疗风险和利益、相关不良反应及禁忌证,包括肿瘤对周围组织的压迫、疾病的严重程度、远期垂体损害的可能性、有生育需求的患者、着重保护垂体功能等。手术作为一线治疗方法,若术后未能治愈,则可考虑药物治疗。如果多巴胺受体激动剂或SSA的最大剂量仍不能有效控制病情,应根据患者症状和生化指标,考虑放疗或再次手术。在选择手术治疗的患者中,可酌情考虑术前先使用SSA治疗12~24周,缩小肿瘤体积以降低手术难度,提高肿瘤全切除的可能性,或改善心脏和呼吸系统的等严重并发症,以创造最优的手术条件并减少手术风险,增加手术成功率。
参考文献
[1]ALEXANDER L,APPLETON D,ROSS WM,et al. Epidemiology of acromegaly in the Newcastle region[J]. Clin Endocrinol(Oxf),1980,12(1):71-79.
[2]BENGTSSON BA,ERNEST I EDEN S,ODEN A,et al. Epidemiology and long-term survival in acromegaly. A study of 166 cases diagnosed between 1955 and 1984[J]. Acta Med Scand,1988,223(4):327-335.
[3]HOSKULDSDOTTIR GT,FJALLDAL SB,SIGURJONSDOTTIR HA. The incidence and prevalence of acromegaly,a nationwide study from 1955 through 2013[J]. Pituitary,2015,18(6):803-807.
[4]AGUSTSSON TT,BALDVINSDOTTIR T,JONASSON JG,et al. The epidemiology of pituitary adenomas in Iceland,1955-2012:a nationwide population-based study[J]. Eur J Endocrinol,2015,173(5):655-664.
[5]GRUPPETTA M,MERCIECA C,VASSALLO J. Prevalence and incidence of pituitary adenomas:a population based study in Malta[J]. Pituitary,2013,16(4):545-553.
[6]TJORNSTRAND A,GUNNARSSON K,EVERT M,et al. The incidence rate of pituitary adenomas in western Sweden for the period 2001-2011[J]. Eur J Endocrinol,2014,171(4):519-526.
[7]MESTRON A,WEBB SM,ASTORGA R,et al. Epidemiology,clinical characteristics,outcome,morbidity and mortality in acromegaly based on the Spanish Acromegaly Registry(Registro Espanol de Acromegalia,REA)[J].Eur J Endocrinol,2004,151(4):439-446.
[8]DAL J,FELDT-RASMUSSEN U,ANDERSEN M,et al. Acromegaly incidence,prevalence,complications and long-term prognosis:a nationwide cohort study[J]. Eur J Endocrinol,2016,175(3):181-190.
[9]GALERNEAU LM,PEPIN JL,BOREL AL,et al. scientific council and investigators of the French national sleep apnoea registry(OSFP).Acromegaly in sleep apnoea patients:a large observational study of 755 patients[J]. Eur Respir J,2016,48(5):1489-1492.
[10]BECKERS A,DALY AF. The clinical,pathological,and genetic features of familial isolated pituitary adenomas[J]. Eur J Endocrinol,2007,157(4):371-382.
[11]CAIMARI F,KORBONIT M. Novel Genetic Causes of Pituitary Adenomas[J]. Clin Cancer Res,2016,22(20):5030-5042.
[12]TRIVELLIN G,DALY AF,FAUCZ FR,et al. Gigantism and acromegaly due to Xq26 microduplications and GPR101 mutation[J]. N Engl J Med,2014,371(25):2363-2374.
[13]SALENAVE S,BOYCE AM,COLLINS MT,et al. Acromegaly and McCune-Albright syndrome[J]. J Clin Endocrinol Metab,2014,99(6):1955-1969.
[14]VORTMEYER AO,GLASKER S,MEHTA GU,et al. Somatic GNAS mutation causes widespread and diffuse pituitary disease in acromegalic patients with McCune-Albright syndrome[J]. J Clin Endocrinol Metab,2012,97(7):2404-2413.
[15]VERGES B,BOUREILLE F,GOUDET P,et al. Pituitary disease in MEN type 1(MEN1):data from the France- Belgium MEN1 multicenter study[J]. J Clin Endocrinol Metab,2002,87(2):457-465.
[16]SYRO LV,SCHEITHAUER BW,KOVACS K,et al. Pituitary tumors in patients with MEN1 syndrome[J]. Clinics(Sao Paulo),2012,67 Suppl 1:43-48.
[17]WU Y,GAO L,GUO X,et al. Pituitary adenomas in patients with multiple endocrine neoplasia type 1:a single-center experience in China[J]. Pituitary,2019,22(2):113-123.
[18]KAMILARIS CDC,FAUCZ FR,VOUTETAKIS A,et al. Carney Complex[J]. Exp Clin Endocrinol Diabetes,2019,127(2-03):156-164.
[19]LS. KIRSCHNER. PRKAR1A and the evolution of pituitary tumors[J]. Mol Cell Endocrinol,2010,326(1-2):3-7.
[20]O’TOOLE SM,DENES J,ROBLEDO M,et al. 15 YEARS OF PARAGANGLIOMA:The association of pituitary adenomas and phaeochromocytomas or paragangliomas[J].EndocrRelat Cancer,2015,22(4):T105-122.
[21]CONSTANTIN T,TANGPRICHA V,SHAH R,et al. Calcium and Bone Turnover Markers in Acromegaly:A Prospective,Controlled Study[J]. J Clin Endocrinol Metab,2017,102(7):2416-2424.
[22]BIHAN H,ESPINOSA C,VALDES-SOCIN H,et al. Long-term outcome of patients with acromegaly and congestive heart failure[J]. J Clin Endocrinol Metab,2004,89(11):5308-5313.
[23]ATTAL P,CHANSON P. Endocrine aspects of obstructive sleep apnea[J]. J Clin Endocrinol Metab,2010,95(2):483-495.
[24]COLAO A,FERONE D,MARZULLO P,et al. Systemic complications of acromegaly:epidemiology,pathogenesis,and management[J]. Endocr Rev,2004,25(1):102-152.
[25]DELHOUGNE B,DENEUX C,ABS R,et al. The prevalence of colonic polyps in acromegaly:a colonoscopic and pathological study in 103 patients[J]. J Clin Endocrinol Metab,1995,80(11):3223-3226.
[26]KATZNELSON L,LAWS ER JR,MELMED S,et al. Acromegaly:an endocrine society clinical practice guideline[J]. J Clin Endocrinol Metab,2014,99(11):3933-3951.
[27]METE O,LOPES MB. Overview of the 2017 WHO Classification of Pituitary Tumors[J]. EndocrPathol,2017,28(3):228-243.
[28]中华医学会内分泌学会,中国垂体腺瘤协作组.中国肢端肥大症诊治指南 [J].中华医学杂志,2013,93(27):2106-2111.
[29]EZZAT S,CASPAR-BELL GM,CHIK CL,et al. Predictive Markers for Post-Surgical Medical Management of Acromegaly:A Systematic Review and Consensus Treatment Guideline[J]. EndocrPract,2019,25(4):379-393.
知识来源
来源:人卫知识数字服务体系
作者:朱惠娟教授,包新杰副教授,中国医学科学院北京协和医院
专家简介
朱惠娟,医学博士,主任医师,教授,博士生导师,中国医学科学院北京协和医院内分泌科常务副主任。1995年毕业于华西医科大学,2008年赴日本熊本大学医学部交流。主持国家自然科学基金等项目,成果发表在JCEM,J Mol Endocrinol,Front Pharmacol。现任中国垂体瘤协作组委员兼秘书,中国医师协会内分泌代谢医师分会青委会副主任委员、中国医师协会青春期医学专业委员会副主任委员、北京医师协会内分泌代谢学专业委员会干事长。主编矮小症、肥胖症以及糖尿病相关书籍5本。2007年获得北京市科委“科技新星”称号。2015年获北京市科学技术奖三等奖(垂体腺瘤的规范化诊治),2016年获华夏医学科技奖二等奖(垂体腺瘤的规范化诊治和垂体功能重建)和北京协和医院“医疗成果奖一等奖”(矮小症的精准诊疗探索)。擅长内分泌代谢疾病的诊治。
包新杰,男,主任医师,博士研究生导师,中国医学科学院北京协和医院神经外科副主任。北京医学会神经外科分会青年委员会副主任委员,中国医促会神经外科分会委员,中国神经科学学会神经外科基础与临床分会委员,北京医师协会神经修复学专业专家委员会委员。研究成果“垂体腺瘤的分子机制研究和临床规范化诊治推广应用”获得中华人民共和国教育部科技进步二等奖和中华医学会科技进步三等奖,“中枢神经系统损伤后神经再生修复的关键技术与机理研究”获得中华人民共和国教育部自然成果奖二等奖。作为首席科学家负责国家重点研发计划“干细胞及转化研究”重点专项;主持国家863课题、国家自然科学基金、北京市自然科学基金、医科院创新工程重大协同创新项目等多项国家级和省部级课题。
- 评价此内容

 3我要打分
3我要打分

 会员登录
会员登录
近期推荐
- 【肺癌进展报告2021】肺癌免疫…
- 【肺癌进展报告2021】放疗联合…
- 【肺癌报告2020】农靖颖教授:…
- 【肺癌报告2020】王汉萍教授:…
- 【肺癌报告2020】杨帆教授:非…
- 【肺癌报告2020】李琳教授:晚…
- 【肺癌报告2020】王志杰教授:…
- 王长利教授:非小细胞肺癌新辅…
- 张力教授:肺癌免疫治疗应用与…
- 赵明教授:肝癌介入治疗研究进展

热门关键词
最新会议
- 2013循证医学和实效研究方法学研讨会
- 欧洲心脏病学会年会
- 世界帕金森病和相关疾病2013年会议
- 英国介入放射学学会2013年第25届年会
- 美国血液学会2013年年会
- 美国癫痫学会2013年第67届年会
- 肥胖学会 2013年年会
- 2013年第9届欧洲抗体会议
- 国际精神病学协会 2013年会议
- 妇科肿瘤2013年第18届大会
- 国际创伤压力研究学会2013年第29届…
- 2013年第4届亚太地区骨质疏松症会议
- 皮肤病协会国际2013年会议
- 世界糖尿病2013年大会
- 2013年国际成瘾性药年会
- 彭晓霞---诊断试验的Meta分析
- 武姗姗---累积Meta分析和TSA分析
- 孙凤---Network Meta分析
- 杨智荣---Cochrane综述实战经验分享
- 杨祖耀---疾病频率资料的Meta分析
友情链接
合作伙伴
Copyright g-medon.com All Rights Reserved 环球医学资讯 未经授权请勿转载!
网络实名:环球医学:京ICP备08004413号-2
关于我们|
我们的服务|版权及责任声明|联系我们
互联网药品信息服务资格证书(京)-经营性-2017-0027
互联网医疗保健信息服务复核同意书 京卫计网审[2015]第0344号
