卢琳、冯铭:一文了解垂体促肾上腺皮质激素细胞腺瘤
库欣综合征又称皮质醇增多症,是由各种病因导致的高皮质醇血症,作用于靶器官引起的以向心性肥胖、高血压、糖代谢异常、低钾血症和骨质疏松为典型表现的一种综合征。据国外文献报告,库欣综合征的年发病率为2/1000 000~10/1000 000,其患病率约40/1000 000。
从病因上分类,库欣综合征可以分为促肾上腺皮质激素(adrenocorticotropin hormone,ACTH)依赖性和ACTH非依赖性,前者包括分泌ACTH的垂体腺瘤和其他部位异位分泌ACTH或促肾上腺皮质激素释放激素(corticotropin-releasing hormone,CRH)的神经内分泌肿瘤,占病因的70%~80%;后者是肾上腺肿瘤(腺瘤和腺癌)或增生自主地分泌过量皮质醇所致,占病因的20%~30%。而垂体性库欣综合征,又称为库欣病(垂体ACTH腺瘤),是库欣综合征中最常见的病因,占患者总数的70%左右。
库欣病除了直接影响糖、脂肪、蛋白质、水电解质等各种物质代谢的平衡,还会影响全身多个系统脏器功能,使机体免疫力下降。如果未得到及时诊治则预后差,严重的低血钾、重症感染及心脑血管并发症可以危及患者生命。库欣病多数为散发,90%是垂体微腺瘤,肿瘤多小于5mm,但有向周边垂体组织浸润的倾向,约10%为大腺瘤。
一、流行病学
库欣病特指由垂体ACTH腺瘤引起的库欣综合征,约占垂体腺瘤的15%,发病率为3~10/(1 000 000•年),好发于25~45岁的女性,男∶女=1∶4~1∶3。但在青春发育前的患者中,好发于男性。在儿童患者群体中,<11岁的垂体腺瘤患儿其库欣病占比为55%。>5岁的儿童库欣综合征患儿中,75%为库欣病。
二、病理生理
1.病因学
垂体腺瘤是良性肿瘤,比较普遍的观点认为垂体肿瘤是单克隆性起源,但长久以来,库欣病的确切发病机制不详。近年来在分子生物学技术飞速进展的基础上,目前研究已经揭示与库欣病发病可能的相关基因包括细胞周期蛋白E1(cyclin E1,CCNE1)、细胞周期蛋白依赖性激酶(cdk5 and abl enzyme substrate 1,CABLES1)、垂体瘤转化基因(pituitary tumor-transforming gene 1,PTTG)、骨形态发生蛋白 4(bone morphogenetic protein 4,BMP4)、SHH(sonic hedgehog)、PAX7(paired box 7)。这些基因涉及细胞周期调控、染色体分离、生长因子调控、细胞分化等方面。此外,miRNA也在疾病发生中有着一定的作用。
尽管多数库欣病患者为散发病例,但有非常罕见的患者为家族性改变,包括多内分泌腺瘤病1型(致病基因为MEN1)、多内分泌腺瘤病2型(致病基因为RET)、多内分泌腺瘤4型(致病基因为CDKN18)、结节性硬化症(致病基因为TSC1/2)。也有关于位于X染色体的剂量敏感的性别反转-先天性肾上腺发育不良基因1(dosage-sensitive sex reversal,adrenal hypoplasia critical region,on chromosome X,gene 1,DAX-1)、DICER-1的胚系突变导致库欣病的报道。截至目前,在库欣病中还没有USP8、PRKAR1A或GNAS的胚系突变的报道。
2.发病机制
阿片黑素促皮质激素原(proopiomelanocortin,POMC)是ACTH的前体,受下丘脑CRH刺激,在垂体产生,可以进一步分解成促黑素、ACTH和β-内啡肽。库欣病患者的垂体加压素V2、V3以及V1b受体均表达上调,因此在CRH和垂体加压素刺激下,库欣病患者的ACTH产生增多。CRH对POMC的调控依赖于多个因子的介导。神经生长因子IB或者Nur77在CRH刺激作用及糖皮质激素负反馈过程中均有参与,其在临床症状显著的患者中明显升高。核受体TR4能够结合POMC启动子并促进转录,TR4通过和糖皮质激素受体相互作用而抑制其减弱POMC表达。
糖皮质激素抵抗是库欣病的一个显著特点。但是编码糖皮质激素受体(glucocorticoid receptor,GR)的NR3C1基因的遗传缺陷在库欣病患者中比较罕见。但有研究发现热休克蛋白90(Heat shock protein 90,HSP90)在库欣病肿瘤组织中过表达;Brg1基因和组蛋白脱乙酰基酶2(HDAC2)参与了染色质重塑调控,这两者在库欣病肿瘤组织中下调;库欣病垂体肿瘤组织中的11β-HSD2和11β-HSD1比例失调,皮质醇被更多地转化为皮质酮从而失活。这些改变损害了糖皮质激素对POMC转录的正常负反馈调控抑制,从而造成糖皮质激素抵抗。
3.泛素特异性蛋白酶8体细胞突变和表皮生长因子受体通路功能改变
通过全外显子组测序的方法,研究发现库欣病肿瘤组织中泛素特异性蛋白酶 8(ubiquitin specific peptidase 8,USP8)的体细胞突变率在35%~67%之间,突变率明显高于之前报道的致病基因。而且USP8的高频突变未在其他类型的垂体腺瘤中出现。
USP8基因表达的是一种大小约130kDa的具有去泛素化酶活性的酶,具有从目标蛋白上切除泛素多肽的功能,这一过程受到14-3-3家族蛋白的调控。USP8的14-3-3蛋白的结合结构域是RSYpSSP,人类中pS为Ser718,这一位点的磷酸化能维持其在胞浆中功能处于未激活状态。一旦这一位点去磷酸化,14-3-3结构域将被释放并激活USP8。
USP8最为重要的功能是对膜蛋白内吞体的调节,特别是以酪氨酸激酶受体(RTK)为主,其影响的底物包括Nrdp1、c-Met、HER2以及表皮生长因子受体(epidermal growth factor receptor,EGFR)。这类膜蛋白被激活后通过内吞以及多泛素化进入细胞内回收,以避免细胞信号通路的过度激活。USP8作用的恰恰是相反的过程,它通过介导去泛素化过程来中断这一过程,从而使受体能够继续激活相应的细胞信号通路。因此,如果USP8被过度激活,与之相关的膜受体的有效作用时间都会相应延长。
EGFR本身参与细胞的生长和增殖,在正常垂体组织中,EGFR存在且处于一个较低水平。但是在mRNA或者蛋白层面,约60%垂体腺瘤中能够检测到EGFR,且库欣病的EGFR表达最高。动物实验证据显示,识别胞内胞外EGFR的抗体在库欣病中高水平表达,而且其分布与ACTH分布一致。同样,EGFR对于POMC(ACTH前体)表达有增强作用。
目前关于USP8在库欣病的发病机制推测为ACTH分泌细胞发生USP8突变后,EGFR持续激活使细胞增殖,激素分泌增多,产生过多的ACTH。USP8突变在女性患者中更为多见,发生率约为男性的两倍,多发于20~40岁的育龄期,而在儿童以及50岁以上患者中USP8则往往是野生型。这一特征可能提示USP8突变和性激素相关。USP8突变对于肿瘤大小的影响尚不明确,不同研究对这一观点也存在分歧。
三、临床表现
库欣病的临床表现是由于ACTH刺激肾上腺产生过多的皮质醇,皮质醇作用于全身靶器官所带来的。大多数库欣病具有显著的功能,然而,20%的库欣病缺乏现实ACTH和皮质醇过多的临床和生化证据,这些肿瘤被称为寂静型促肾上腺皮质激素腺瘤,通常因为垂体意外瘤或当肿瘤引起神经或眼科症状时,包括急性出血坏死导致的卒中而被发现。
1.脂肪代谢紊乱和分布异常
皮质醇可以引起脂肪的异常分布,因此患者呈明显向心性肥胖,满月脸、水牛背、锁骨上脂肪垫、悬垂腹、四肢相对瘦小、体重轻度或中度增加。
2.蛋白质代谢异常
过量皮质醇促使蛋白质的分解代谢大于合成代谢,出现负氮平衡,导致皮肤菲薄,结缔组织减少,毛细血管扩张,呈多血质,皮肤出现紫纹。毛细血管脆性增加,易出现皮下瘀斑。肌无力,肌萎缩,皮肤破溃不易愈合、易感染。患者的免疫力下降。
3.对钙磷代谢的影响
高皮质醇血症抑制肠道对钙的吸收,同时因为骨钙被动员,尿钙排泄增多,易形成泌尿系结石。此外,在负氮平衡的共同作用下,骨质疏松高发,可引起脆性骨折,骨折的部位以肋骨骨折、椎体压缩性骨折多见。
4.糖代谢异常
过量皮质醇抑制糖利用,促使肝脏糖异生,并产生胰岛素抵抗,可以导致糖耐量减低(50%~75%)和糖尿病(8%~20%)。
5.水电解质代谢紊乱
皮质醇增多可以竞争性作用于肾小管的盐皮质激素受体,发挥潴钠排钾作用,临床上可表现为低血钾、低血氯、严重水电解质代谢紊乱可致低钾性碱中毒,需急诊处理。水钠潴留可致高血压,发生率为80%~90%。
6.性腺功能异常
过多皮质醇抑制垂体促性腺激素的释放,71%~87%的女性患者有性欲减退、月经稀少、不规则或闭经、溢乳、不孕;约20%的男性患者性欲减退、阳痿、精子减少、睾丸萎缩。此外,因为ACTH分泌的增加,刺激肾上腺网状带产生脱氢表雄酮增加,无论性别,男女均可表现为皮肤痤疮(多见于面部和胸背部)、毳毛增多。
7.其他表现
有的患者精神异常,可表现为情绪不稳、烦躁、类偏狂、类精神分裂等,但以忧郁症多见。青春期前发病者由于过量皮质醇抑制GH分泌,会严重影响生长发育。垂体ACTH大腺瘤可有压迫症状,可有视力下降、视野缺损、头痛等症状。
8.皮质醇增多
皮质醇增多所带来的血脂升高、糖代谢异常和高血压可导致血管粥样硬化,血管平滑肌及内皮细胞增殖,故晚期库欣病患者常并发心血管、脑血管疾病。晚期库欣病患者多因心脑血管疾病及感染性疾病而死亡。
四、病理诊断
根据2017年WHO病理对垂体疾病的分类,库欣病,即ACTH腺瘤是一种表达ACTH和其他POMC相关肽的垂体腺瘤,起源于TPIT谱系的腺垂体细胞。在组织学上可分为三种亚型:致密颗粒型ACTH腺瘤、稀疏颗粒型ACTH腺瘤和Crooke细胞腺瘤。最常见的组织类型为致密颗粒型ACTH腺瘤。
与其他垂体腺瘤一样,ACTH腺瘤大体表现为组织质软,呈现白色至灰红色。光镜下肿瘤细胞表现为嗜碱性,PAS反应阳性。无论有无分泌功能和哪一种组织亚型,ACTH腺瘤的免疫组化共同特征为弥漫性染色阳性的TPIT、NeuroD1和低分子量细胞角蛋白(CAM5.2)。致密颗粒型ACTH肿瘤细胞呈片状生长,由嗜碱性细胞构成,细胞胞浆内见丰富的PAS染色阳性和ACTH染色强阳性。稀疏颗粒型肿瘤细胞胞浆较少,呈弱嗜碱性或嫌色性,PAS和ACTH染色呈现弱阳性。Crooke细胞腺瘤由Crooke透明样变的腺瘤细胞组成,表现为低分子量角蛋白环状表达,ACTH阳性表达颗粒分布于细胞膜周围和核旁。
寂静型ACTH腺瘤(silent corticotroph adenoma,SCA),临床常表现为无功能的肿瘤。可分为两个亚型:1型(致密颗粒型)和2型(稀疏颗粒型)。和1型相比,SCA 2型反映侵袭性、迁移和增殖的生物标志表达更高,如成纤维细胞生长因子受体4(fibroblast growth factor receptor 4,FGFR4),基质金属蛋白酶 1(matrix metalloproteinase-1,MMP1)和CD29(也称为Integrin β1)。罕见情况下,SCA腺瘤组织缺乏ACTH的表达,但表达TPIT。
ACTH腺瘤的增殖活性变异很大。p27的下调表达很常见;与功能性肿瘤不同的是,在SCA组织中半乳糖凝集素3(galectin 3)为弱表达或无表达。也有研究提示galectin 3在侵袭性ACTH腺瘤中存在表达。
只有在发生远处转移或脑脊髓播散才可以确诊垂体ACTH腺癌。大多数垂体ACTH腺瘤是孤立的病灶,但少部分是双腺瘤的组分之一,还有一些罕见病例ACTH腺瘤组织中混杂有神经节细胞瘤组织或肾上腺皮质迷芽瘤(adrenal cortical choristoma)。
对肿瘤组织周围的腺垂体细胞的评估也很重要,以明确ACTH细胞是否存在Crooke透明样改变。这个形态学改变反映了下丘脑-垂体-肾上腺轴存在高皮质醇血症,在类库欣患者和SCA患者中看不到这种变化。
五、定性诊断
库欣综合征的定性诊断主要包括临床表现和生化检测。临床表现包括满月脸、水牛背、多血症、皮肤变薄和/或皮肤紫纹、高血压、糖尿病、与年龄不相称骨质疏松等特征性的临床表现。生化检测主要包括如下:
(一)疑诊库欣综合征的筛查试验
1.24小时尿游离皮质醇
皮质醇在体内与皮质醇结合球蛋白(cortisol binding globulin,CBG)结合,因此测定的血皮质醇主要为总皮质醇,易受改变CBG水平的生理或病理状态影响。而24小时尿游离皮质醇(urine free cortisol,UFC)测定的是游离皮质醇,故不受CBG的浓度影响。24小时UFC超过正常上限即判断为阳性,诊断CS的敏感性可达到91%~96%。
在进行24小时UFC的结果解读时,也需要注意有些因素会影响24小时UFC结果判断:①在中、重度肾功能不全患者,GFR < 60 mL/min/1.73m²时可出现UFC明显降低的假阴性结果;②饮水过多(≥5L/d),任何增加皮质醇分泌的生理或病理应激状态都会使UFC升高而出现假阳性结果;③检测方法的影响,目前采用最多的化学发光法和放射免疫分析法(RIA),可受合成糖皮质激素或皮质醇代谢产物交叉反应的影响;④基于分子结构的高效液相色谱串联质谱分析法(LC-MS/MS)虽然可避免上述问题,但会受某些药物(如:卡马西平和非诺贝特)干扰测定而使结果假性升高;此外,应用LC-MS/MS测定尿皮质醇浓度较RIA法测定低40%。
2.午夜血清/唾液皮质醇测定
体皮质醇分泌呈现明显的昼夜节律,血皮质醇水平在午夜达最低值。唾液皮质醇与血皮质醇可以达到动态平衡,反映体内游离皮质醇的状态,与血皮质醇类似,CS患者血清午夜血皮质醇低谷会消失。抑郁症、酗酒、肥胖和糖尿病患者的HPA轴活性增强,而地塞米松抑制试验(dexamethasone suppression test,DST)较单次测定血、唾液或尿皮质醇更有意义。如进行午夜血清皮质醇测定,应尽量保证采血时处于睡眠状态,为了避免采血带来的应激,可采取提前静脉置管的操作。判断切点:诊断CS的午夜血清皮质醇值≥50nmol/L(1.8µg/dl),敏感性达100%,但特异性仅20%。清醒状态下血清皮质醇值≥207nmol/L(7.5µg/dl),诊断的敏感性>96%,特异性87%。唾液中皮质醇呈游离状态,其浓度与血中游离皮质醇浓度平行。各国文献报道测定午夜唾液皮质醇用于诊断CS的敏感性为92%~100%、特异性为93%~100%。
3.1mg过夜地塞米松抑制试验
1mg过夜地塞米松抑制试验(overnight dexamethasone suppression test,ODST)的做法为午夜11~12点口服地塞米松1mg,次日晨8:00采集服药后血皮质醇标本。服药后血清皮质醇值≥50nmol/L(1.8µg/dl)为不抑制,诊断CS的敏感性>95%、特异性约80%;若提高切点至 140nmol/L(5µg/dl),其敏感性为91%,特异性可提高至>95%,但敏感性降低。
在该项检查中,需注意患者对地塞米松的吸收和代谢率不同可影响DST的结果;部分药物如苯巴比妥、卡马西平和利福平等可通过诱导CYP3A4加速清除地塞米松的清除而导致假阳性;而肝、肾衰竭患者的地塞米松清除率降低可以导致假阴性。
4.经典小剂量地塞米松抑制试验
经典小剂量地塞米松抑制试验(low dose dexamethasone suppression test,LDDST,地塞米松用法 2mg/d×48h)检查前留24小时UFC或者清晨血皮质醇作为对照,之后开始口服地塞米松每次0.5mg,间隔6小时服用一次,连续2天,在服药的第2日再留24小时UFC水平或服药2天后测定清晨血皮质醇水平,若UFC未能下降到正常值下限以下或服药后血皮质醇≥1.8µg/dl,为经典小剂量DST不被抑制。两者的敏感性和特异性相差不大,均可达到敏感性> 95%。
虽然24小时UFC是经典的LDDST的主要判断标准,但因采集标本不易,而血皮质醇无论是留取标本还是测定都更为简单,目前是依据24小时UFC还是血皮质醇来做为判断标准,各家医学中心做法不一。北京协和医院选择有病理证实明确诊断为CS的病例67例,包括ACTH依赖性CS 60例(库欣病53例,异位ACTH综合征7例)和非ACTH依赖性CS 7例,所有患者在采集24小时UFC的时候同步采集清晨血皮质醇样本,结果提示以24小时UFC作为判断标准,LDDST中诊断符合率92.54%;若以清晨血皮质醇<1.8μg/dl、<4.0μg/dl、<5.0μg/dl和<对照值的50% 为标准,则诊断符合率分别为97.01%、86.57%、83.58%和71.15%。因此,在LDDST中,采用服药后清晨血皮质醇<1.8μg/dl的切点其敏感性最高。而其他两种切点(血皮质醇<5μg/dl和血皮质醇<对照值的50%)在诊断CS的符合率方面,因敏感性过低,不推荐用于CS的定性诊断。
(二)库欣综合征的定位实验室检查
定位的实验室检查包括血ACTH的测定和大剂量地塞米松抑制试验。
1.血促肾上腺皮质激素测定
清晨8:00采血,因ACTH的半衰期很短,取血后需要将血标本冰浴,并尽快低温离心测定。通常认为如血ACTH<2.2pmol/L(10pg/ml),则考虑 ACTH 非依赖性 CS,如ACTH>4.4pmol/L(20pg/ml),则考虑为ACTH依赖性CS。
2.经典大剂量地塞米松抑制试验
经典大剂量地塞米松抑制试验(high dose dexamethasone suppression test,HDDST,地塞米松用法 8mg/d × 48h)检查前留24小时UFC或血皮质醇作为对照,之后口服地塞米松每次2.0mg,间隔6小时服用一次,连续2天,在服药的第2日再留24小时UFC或服药2天后测定清晨血皮质醇,若UFC或者血皮质醇下降到对照值的50%以下为经典大剂量DST被抑制,支持库欣病的诊断。该试验鉴别库欣病与异位ACTH综合征的敏感性为60%~80%,特异性80%~90%。
3.联合法小剂量及大剂量地塞米松抑制试验
LDDST和HDDST也可以连续进行,试验第1,2天连续2天留24小时尿测定UFC水平,取2天结果的平均值作为对照值。第3~4天连续口服地塞米松每次0.5mg,间隔6小时服用一次,第4天留24小时尿测定UFC水平,第5~6天连续口服地塞米松每次2.0mg,间隔6小时服用一次,第6天留24小时尿测定UFC水平。第4天的UFC水平若下降到正常值下限以下(标准同上)为LDDST被抑制,反之则为LDDST不被抑制;第6天的24小时UFC水平下降到对照平均值的50%以下称为HDDST被抑制,反之则为大剂量DST不被抑制。
联合法与经典法DST比较,前者可以连续进行,更为节约时间,而后者需要LDDST和HDDST分开进行。关于其诊断效力是否相同,北京协和医院回顾性分析了152例经手术病理证实的CS病例,比较了经典法与联合法两种DST做法与最终诊断的符合率。结果显示经典法及联合法之HDDST对ACTH非依赖性CS诊断的符合率分别为94.2%和95.5%,两组之间无显著性差异(P = 0.83)。经典法分别行LDDST及HDDST和联合法DST诊断库欣病的敏感性分别为81.5%和77.8%,特异性分别为92.5%和95.5%。因此联合法小剂量及大剂量DST比经典法操作简便,节省时间,可用于CS的定性、定位诊断。
六、定位诊断
库欣综合征可以由垂体ACTH分泌瘤、肺/纵隔内的异位ACTH分泌瘤和肾上腺肿瘤自主分泌皮质醇引起。鞍区磁共振成像(magnetic resonance imaging,MRI)是诊断库欣病的首选方法,而CT是异位ACTH分泌瘤和肾上腺肿瘤的首选检查方法。正电子发射断层/计算机断层显像(positron emission tomography/CT,PET/CT)可能在异位病灶的检出和残存、复发病灶的判断方面具有独特的价值。近年来发展的正电子发射断层/磁共振显像(positron emission tomography/MRI,PET/MRI)技术对于隐匿性或术后复发的库欣病垂体瘤病灶的检出具有重要的价值。
1.鞍区磁共振成像
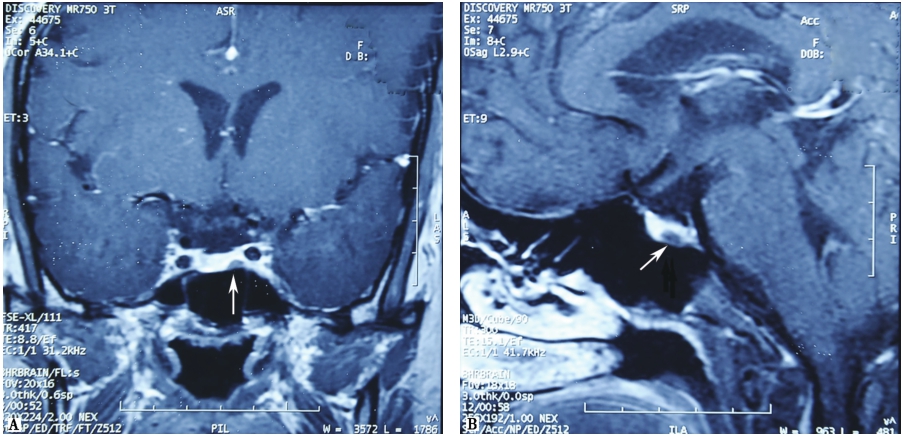
库欣病多为垂体微腺瘤,常需要进行鞍区动态增强MRI,以提高肿瘤检出率。在动态增强中,微腺瘤的强化慢于且弱于正常垂体(图1),因此在增强早期可形成较好的对比。垂体微腺瘤的间接征象包括:垂体形态不对称、信号不均、垂体柄偏移、鞍底倾斜凹陷等。
当鞍区动态增强MRI检查阴性时,要考虑到肿瘤极其微小,未达到目前MRI的空间分辨率的可能。因此进一步可行双侧岩下窦静脉取血明确诊断。同时,还应考虑异位ACTH综合征的可能。故还需要进一步行胸部CT、腹盆部CT检查等。
2.胸部CT
约90%的异位ACTH肿瘤在肺或纵隔内,随着检查技术手段的进步,胸部CT增强扫描影像学检查有助于发现临床疑诊异位ACTH综合征的胸部原发肿瘤。如考虑可能存在其他异位病灶,则根据情况选择腹部、胰腺或盆腔的CT检查来探查病灶。
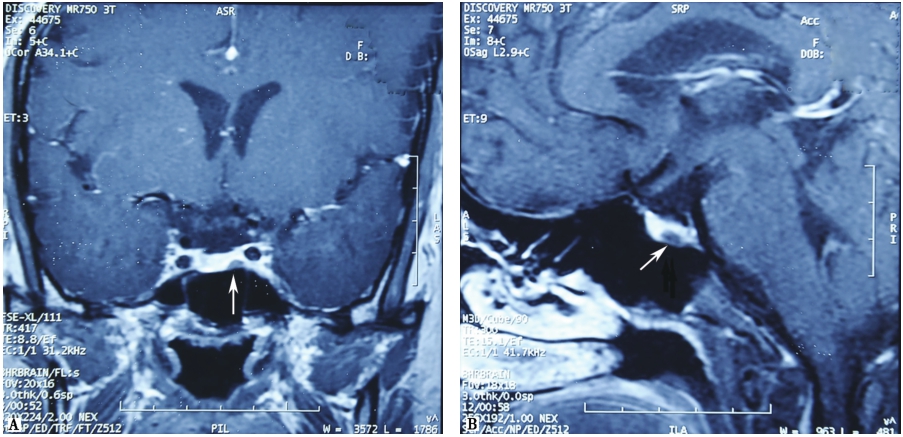
图1 A.垂体T1增强冠状位图像,显示垂体左翼低强化区为垂体微腺瘤;B.垂体T1增强矢状位图像。图中白色箭头所指为垂体微腺瘤
3.双侧肾上腺影像学
肾上腺影像学检查方法包括B超、CT、MRI等检查,对诊断ACTH非依赖性CS患者有很重要的意义。推荐首选双肾上腺薄层增强CT扫描(层厚2~3mm),有条件可同时行三维重建以更清晰地立体显示肾上腺病变的形态。
4.双侧岩下窦静脉取血+去氨加压素兴奋试验
ACTH依赖性CS如临床、生化、影像学检查结果不一致或难以鉴别病因时,建议行双侧岩下窦静脉取血(bilateral inferior petrosal sinus sampling,BIPSS)以鉴别ACTH来源于垂体还是非垂体,为了增加该检查的准确性,还需要同时进行CRH或去氨加压素(D-amino D-arginine vasopressin,DDAVP)兴奋试验。BIPSS是有创性血管内介入检查,操作需要一定的技巧,故建议在经验丰富的医疗中心进行。操作方法为经股静脉插管至双侧岩下窦后,可应用数字减影血管成像术证实插管位置是否正确和岩下窦解剖结构是否正常。岩下窦(inferior petrosal sinus,IPS)与外周(peripheral,P)血浆ACTH比值在基线状态≥2和/或CRH/DDAVP刺激后≥3则提示库欣病。
BIPSS应在患者皮质醇水平升高提示肿瘤活跃分泌ACTH时进行检查,避免在周期性库欣静止期进行。技术因素的影响和静脉回流的异常可导致库欣病患者出现假阴性结果。在经验丰富的医疗中心,BIPSS诊断库欣病的敏感性为95%~99%,特异性为95%~100%,术后严重并发症如深静脉血栓、下肢静脉栓塞、肺栓塞、蛛网膜下腔出血等少见。采用BIPSS联合CRH/DDAVP刺激试验具有是确诊库欣病的金指标,但对垂体微腺瘤的左右侧定位意义有限。
5. 18F-FDG PET/CT检查
正电子发射断层/计算机断层显像18F-FDG PET/CT检查时正常垂体位于本底较低的鞍区,垂体腺瘤对18F-FDG的摄取高于周围正常垂体组织。所以尽管空间分辨率有限(4~6mm不等),18F-FDG PET/CT仍可能发现CT、MRI难以检出的垂体微腺瘤。因此,在怀疑库欣病而其他检查无阳性发现或不确定时,或在术后复发而CT、MRI很难与术后改变区分时,可考虑选用18F-FDG PET/CT显像。
6.生长抑素受体显像
生长抑素受体(somatostatin receptor,SSTR)是位于细胞膜表面的G蛋白耦联受体,有5种亚型,分别是SSTR1、SSTR2、SSTR3、SSTR4、SSTR5。而异位分泌ACTH的神经内分泌肿瘤组织高度表达SSTR2,将放射性核素标记的奥曲肽引入体内,能与肿瘤细胞表面的SSTR2特异性、高亲和力结合,使异位肿瘤显像。以99mTc-OCT为示踪剂的单光子发射计算机断层(single photon emission tomography,SPECT)显像对于寻找异位ACTH综合征的病灶具有一定的价值,但敏感性较低,仅为49%,结果如为阴性也不能排除异位ACTH综合征,还需要进行其他影像学检查进行病灶的定位。
7.正电子发射断层/磁共振显像
目前PET/MRI可以用两种核素标记,分别为18F-FDG和68Ga-DOTATATE,可用于区分影像学不明确或者肿瘤复发无法区分肿瘤与瘢痕组织的情况。北京协和医院相关研究提示在16例接受首次手术的垂体腺瘤进行18F-FDG-PET/MRI,其SUVmax为6.8 ± 3.7,明显高于正常垂体组织(3.2 ± 1.1)。而同时进行68Ga-DOTATATE-PET/MRI,则腺瘤表现为中度摄取,SUVmax为3.8 ± 2.6,低于正常垂体组织(SUVmax,6.2 ± 3.2)。在 11例疑似复发性垂体瘤中,18F-FDG-PET/MRI的 SUVmax 为 6.1 ± 3.5,显著高于周围正常垂体组织(2.5 ± 1.1)。而区分腺瘤和垂体组织最好的判断标准为18F-FDG/68Ga-DOTATATE-PET/MRI的SUVmax比值,其次为单独应用18F-FDG-PET/MRI,其切点分别为1.04和3.88。因此PET/MRI是一种功能性垂体瘤的理想的诊断工具,双示踪剂18F-FDG和68Ga-DOTATATE PET/MRI可以用于垂体微腺瘤与正常垂体组织的区别。但还需要更多的数据以支持PET/MRI在垂体腺瘤中的应用。
七、治疗
(一)治疗目标
治疗原发病、降低皮质醇水平、缓解临床症状体征、治疗相关系统的并发症、保护垂体功能、提高生活质量。
(二)治疗方法
1.手术治疗
手术是垂体ACTH腺瘤患者的首选治疗方法。
(1)术后疗效判断:
库欣病经蝶窦入路手术早期术后缓解率为65%~98%,长期随访中肿瘤复发率为2%~35%。对于首次治疗未缓解的患者,再次手术能够使37%~61%的患者达到缓解,但可能增加脑脊液漏及垂体功能低下的风险。患者随访0.3~37年后发现7%~34%出现肿瘤复发,复发部位常位于原发部位或相邻部位。
术后一周内清晨血清皮质醇测定是目前公认的用于评估疗效的指标。目前多数学者认为血清皮质醇水平低于56nmol/L(2µg/dl)或140nmol/L(5µg/dl)者为缓解。24小时UFC可作为辅助评估工具,其低于28~55nmol(10~20μg)提示缓解,24小时UFC 高于 276nmol/L(100µg)则提示肿瘤残存。
(2)围术期糖皮质激素替代:
术前、术中不需要使用糖皮质激素,但如患者出现血压下降,不明原因发热、低钠血症等肾上腺皮质功能减退表现,尽可能先抽血留取皮质醇、ACTH血样标本后,再补充糖皮质激素,建议给予静脉输注氢化可的松100~200mg,并逐渐替代为口服糖皮质激素。
术后3天内监测清晨血清皮质醇。如果血清皮质醇< 55nmol/L(2µg/dl)时,需立即补充糖皮质激素;如果血清皮质醇55~276nmol/L(2~10µg/dl)时,结合患者的症状,如患者出现烦躁、低血压,恶心呕吐、食欲差、不明原因发热、低钠血症等肾上腺皮质功能减退表现,补充糖皮质激素;如血清皮质醇 > 276nmol/L(10µg/dl),不需要立刻补充糖皮质激素,但要个体化地根据患者是否出现肾上腺皮质功能减退症状来决定是否补充。
(3)复发库欣病的外科处理:
术后鞍区正常解剖结构紊乱,术野内疤痕形成,不易分辨肿瘤和垂体组织,给再次经蝶窦入路手术带来困难。手术过程中鞍底位置判断困难时,可借助术中X线监测、神经导航、术中MRI等手段寻找鞍底,对侵袭海绵窦的肿瘤术中超声对识别颈内动脉有参考价值。
对于临床症状和内分泌检查均支持肿瘤复发,但MRI阴性者,需根据术者经验和手术条件做出综合判断,决定是否进行垂体探查术;术中发现明确肿瘤者,应行肿瘤切除加瘤周垂体大部分切除;如果术中未能见到明确肿瘤,可根据BIPSS结果对ACTH优势侧进行垂体大部分切除;若BIPSS未提示ACTH优势侧,可行初次肿瘤侧垂体大部分切除。经蝶窦入路垂体腺瘤切除加瘤周垂体组织切除是治疗复发性库欣病的首选方法。
2.药物治疗
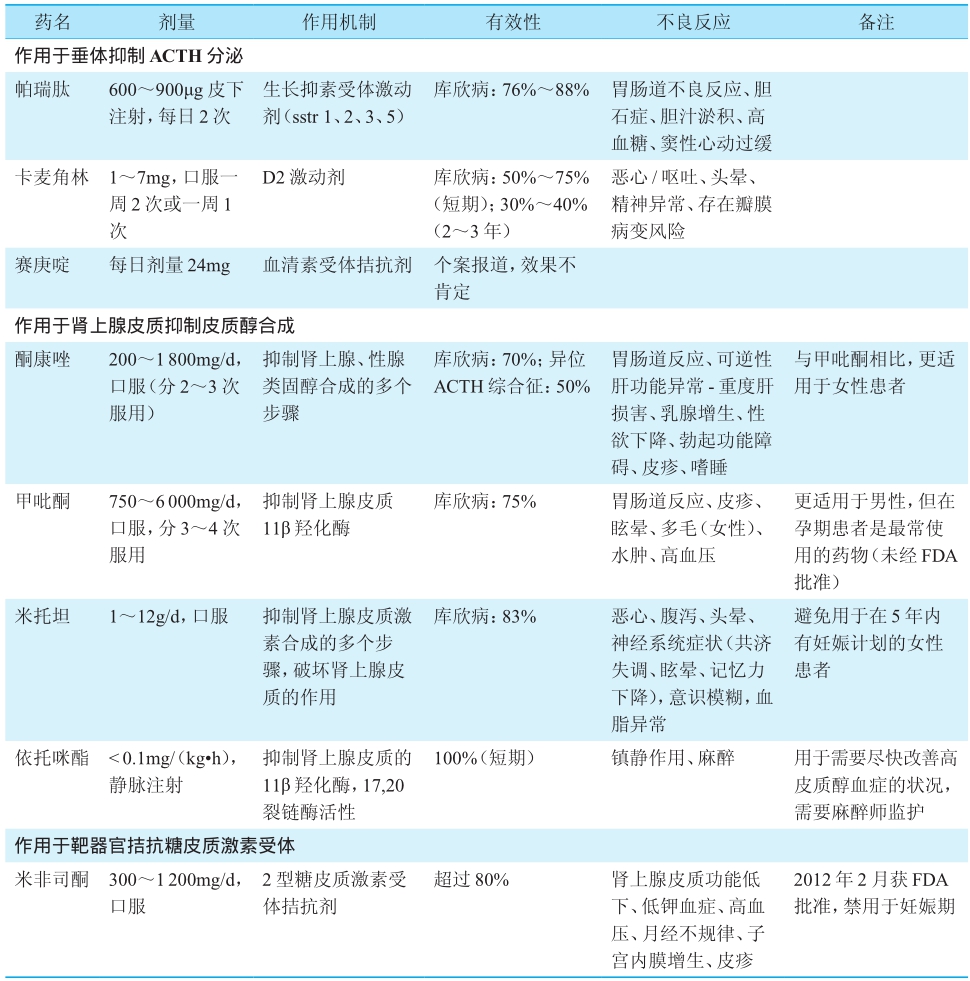
国内治疗库欣病的有效药物不多,临床证据多数来源于小样本、回顾性、单中心研究,总体疗效不佳,因此药物治疗处于辅助地位,适应证为:不适合手术、已经接受了放疗但尚未起效的患者,且一般情况不适宜行双侧肾上腺切除者;严重高皮质醇血症、出现急性精神病、高血压、严重感染等情况时需要及时降低皮质醇水平,为进一步手术创造机会的患者。可以用于库欣病治疗的药物特点总结于表1。
表1 可用于治疗库欣病的药物用法及特点
3.放射和放射外科治疗
放射治疗通常不作为库欣病的首选治疗方法。对术后完全缓解的患者不推荐预防性放疗,但对术后病理为“不典型垂体腺瘤”的患者建议术后放疗以减少复发机会。适应证:①手术残留和/或复发的库欣病;②不适宜/不接受手术的垂体微腺瘤患者;③复发的侵袭性、垂体癌的辅助治疗;④Nelson综合征。
(1)常规放射治疗:
库欣病分次放疗可以选用常规照射技术、三维适形放疗技术及调强放疗技术。随着放疗技术的进步,推荐采用三维适形放疗技术或调强放疗技术进行精确放疗。以MRI和CT图像融合确定照射区及周围可能累及器官,精准评估剂量分布。照射总剂量建议45~50Gy,常规分割20~25次。常规分次放疗库欣病起效缓慢,起效时间一般为6个月至2年,生化缓解率42%~83%,肿瘤控制(影像学上肿瘤体积稳定或缩小)率93%~100%。
最常见并发症为腺垂体功能低下,发生率约为19%~25%,少见的并发症为视路损伤,约0.8%。
(2)伽玛刀放射外科:
伽玛刀放射外科(Gamma knife radiosurgery,GKS)控制垂体腺瘤生长的边缘处方剂量12~16Gy;达到生化指标缓解的处方剂量18~35Gy。建议采用平均25Gy的周边剂量。生化缓解率40%~80%,肿瘤控制率91%~100%,平均缓解时间为10~25个月。
并发症:①腺垂体功能低下最常见,发生率为23%~31.5%,发生高峰为治疗后的4~8年;②视神经和海绵窦内脑神经的损伤比例为4%~5.2%。
4.双侧肾上腺全切术
其原理是切除ACTH的靶器官从而彻底缓解高皮质醇血症。但患者必须终身服用激素替代治疗,并且在某些应激状态下可能导致肾上腺皮质危象。
双侧肾上腺全切除后,缺乏皮质醇对下丘脑的负反馈作用,可能会使垂体肿瘤生长,增大的肿瘤压迫垂体导致垂体功能减退,及ACTH分泌增多而出现皮肤色素沉着等症状称为Nelson综合征,发生率为21%(0%~47%)。双侧肾上腺切除后宜严密监测血浆ACTH水平和垂体MRI,如影像学发现垂体肿瘤则应手术切除或放射治疗。
知识来源
来源:人卫知识数字服务体系
作者:卢琳,冯铭,中国医学科学院北京协和医院
专家简介
卢琳
博士学位。1999年毕业于中国协和医科大学,获博士学位,之后在北京协和医院内分泌科工作至今,历任住院医师、住院总医师、主治医师、副主任医师,主任医师。曾在University of Michigan进行学习交流。多次在国内外内分泌学术会议上进行学术交流。擅长内分泌代谢疾病的诊治。
冯铭
医学博士,副主任医师,博士研究生导师,留美博士后。
历任神经外科住院医师、主治医师、副主任医师。2022年获得国家卫健委主任医师资格。 作为访问学者在美国Johns-Hopkins、MGH、Cedars-Sinai医学中心参观。负责中国垂体疾病注册中心日常工作。主持5项国家及省部级课题,发表50余篇文章,授权专利2项,软件著作权2项。曾获中华医学科技奖等5项奖励。
- 评价此内容

 3我要打分
3我要打分

 会员登录
会员登录
近期推荐
- 【肺癌进展报告2021】肺癌免疫…
- 【肺癌进展报告2021】放疗联合…
- 【肺癌报告2020】农靖颖教授:…
- 【肺癌报告2020】王汉萍教授:…
- 【肺癌报告2020】杨帆教授:非…
- 【肺癌报告2020】李琳教授:晚…
- 【肺癌报告2020】王志杰教授:…
- 王长利教授:非小细胞肺癌新辅…
- 张力教授:肺癌免疫治疗应用与…
- 赵明教授:肝癌介入治疗研究进展

热门关键词
最新会议
- 2013循证医学和实效研究方法学研讨会
- 欧洲心脏病学会年会
- 世界帕金森病和相关疾病2013年会议
- 英国介入放射学学会2013年第25届年会
- 美国血液学会2013年年会
- 美国癫痫学会2013年第67届年会
- 肥胖学会 2013年年会
- 2013年第9届欧洲抗体会议
- 国际精神病学协会 2013年会议
- 妇科肿瘤2013年第18届大会
- 国际创伤压力研究学会2013年第29届…
- 2013年第4届亚太地区骨质疏松症会议
- 皮肤病协会国际2013年会议
- 世界糖尿病2013年大会
- 2013年国际成瘾性药年会
- 彭晓霞---诊断试验的Meta分析
- 武姗姗---累积Meta分析和TSA分析
- 孙凤---Network Meta分析
- 杨智荣---Cochrane综述实战经验分享
- 杨祖耀---疾病频率资料的Meta分析
友情链接
合作伙伴
Copyright g-medon.com All Rights Reserved 环球医学资讯 未经授权请勿转载!
网络实名:环球医学:京ICP备08004413号-2
关于我们|
我们的服务|版权及责任声明|联系我们
互联网药品信息服务资格证书(京)-经营性-2017-0027
互联网医疗保健信息服务复核同意书 京卫计网审[2015]第0344号
