24岁男青年嗅觉障碍伴第二性征不发育 病因何处寻?
24岁青年男性,嗅觉障碍伴第二性征不发育和男性乳房发育。如何进行功能和病因诊断?患者是否有生育机会?如何选择治疗方案?是否会遗传给下一代?
【病例简介】
患者,男,24岁。因“第二性征不发育”于2016年5月入院。患者足月分娩,足先露,出生体重2kg,Apgar评分不详,母乳喂养;年幼时挑食,力气较同龄人小。学习成绩一般,学历中专。18岁时因身材矮小第二性征不发育就诊于当地男科医院,给予肌内注射药物治疗(具体药名不详),治疗3个月,因症状无明显改善而停用。21岁发现双侧乳腺稍增大,偶有发胀,无疼痛,未就诊。本次就诊于华山医院内分泌科门诊,查LH 0.42IU/L,FSH 0.66IU/L,T 0.4nmol/L,E2 23.2pmol/L,PRL 5.6ng/ml,TSH 2.373mIU/L,FT3 5.53pmol/L,FT4 10.9pmol/L,皮质醇(早上8:00)14.17μg/dl。考虑低促性腺激素性性腺功能减退症,为进一步诊治收住内分泌科。追问病史,患者自幼有嗅觉障碍。
家族史:否认性发育不良家族史。父母性发育正常,父亲身高175cm,母亲身高168cm。
入院查体:体温37℃,脉搏84次/分,呼吸20次/分,血压122/78mmHg,身高172cm,体重56kg;神志清楚,营养中等,智力正常,声细,皮肤细嫩,腋毛数根。Tanner生殖器发育及阴毛Ⅱ期。嗅觉:不能区分酒精、白醋和水。四肢无畸形。
【实验室及辅助检查】
血ACTH(早上8:00)14.90pg/ml,血皮质醇(早上8:00)14.17μg/dl。
TSH 2.39mIU/L,TT3 1.86nmol/L,TT4 80.50nmol/L,FT3 5.30pmol/L,FT4 13.34pmol/L。
GH 0.2mIU/L,IGF-1 190μg/L。
LH 0.33IU/L,FSH 0.67IU/L,T 0.34nmol/L↓。
PRL 5.6ng/ml,E2 23.2pmol/L,P 1.35nmol/L。
垂体MRI增强(图1):未见明显异常。
骨龄片:左侧桡骨、尺骨远端及第2~5掌骨远端骨骺未愈合,骨龄约17岁。
睾丸B超:双侧睾丸小(右侧21mm×8mm,左侧20mm×8mm,睾丸体积指数=1.64ml),附睾薄。精索静脉未见扩张。[注:睾丸体积指数=(右侧睾丸长×宽+左侧睾丸长×宽)/2]


图1 垂体MRI增强
A.冠状位;B.矢状位
【内分泌功能试验】
戈那瑞林(GnRH)兴奋试验:
L H峰值8.46IU/L(60min),详见图2。

图2 戈那瑞林兴奋试验
戈那瑞林(GnRH)延长兴奋试验(连续静脉注射戈那瑞林100μg qd×5天,第5剂注射前、注射后多点采血测定LH和FSH):
LH峰值15.04IU/L(30min),FSH峰值7.48IU/L(60min),详见图3。
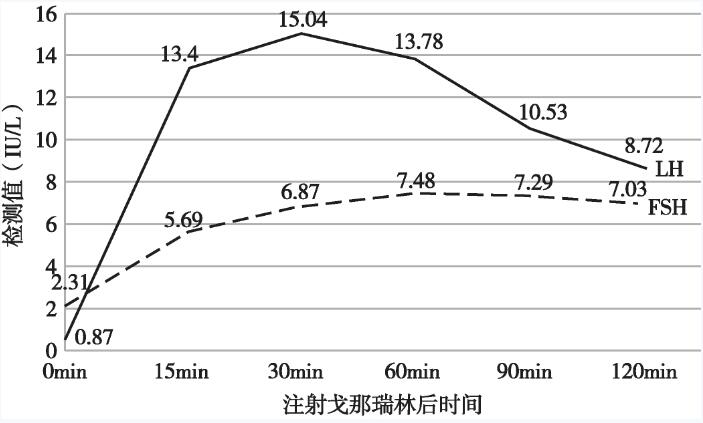
图3 戈那瑞林延长兴奋试验
【诊治经过】
患者男性,24周岁,自幼嗅觉障碍,查体见生殖器发育及阴毛TannerⅡ期,睾丸体积小,提示男性第二性征发育不良;查LH 0.33IU/L,FSH 0.67IU/L,T 0.34nmol/L,戈那瑞林兴奋试验中LH峰值8.46IU/L(60min),提示性腺功能减退的定位在下丘脑,垂体促性腺激素分泌功能良好;结合GH、IGF-1、ACTH、皮质醇、PRL、甲状腺功能等其他垂体及靶腺功能均在正常范围内,鞍区MRI未见明显异常,考虑伴有嗅觉障碍的特发性低促性腺激素性性腺功能减退症,即Kallmann综合征。
患者现24岁,有生育需求,身高在同龄人平均范围,无继续增高需求,结合戈那瑞林延长试验LH、FSH对GnRH反应良好,给予GnRH泵治疗(10μg/90min),门诊随访。
【随访与转归】
2016年6月(GnRH泵治疗1个月):FSH 8.01IU/L,LH 18.01IU/L,T 16.99nmol/L。
2016年11月(GnRH泵治疗半年):FSH 4.30IU/L,LH 6.66IU/L,T 26.68nmol/L。胡须、腋毛生长情况如成年人;生殖器阴毛发育Ⅳ期。身高未再增长。
2017年4月(GnRH泵治疗1年):FSH 3.45IU/L,LH 6.68IU/L,T 16.67nmol/L。
精液检查:精子计数26×109/ml(参考范围:>20×109/ml),正常形态21%(参考范围:80%),精子活力A+B级25%(参考范围:≥50%);有待随访。
【经验与体会】
(一) IHH的诊断和鉴别诊断
特发性低促性腺激素性性腺功能减退症(idiopathic hypogonadotropic hypogonadism,IHH)的诊断依赖症状、体征、性激素测定、GnRH兴奋试验及相关影像学检查等。诊断可分为3步:定性诊断、定位诊断及病因诊断。男性骨龄>12岁或生物学年龄>18岁,而无第二性征出现和睾丸增大,睾酮水平低于3.47nmol/L,可定性为性腺功能减退。LH、FSH降低(或“正常”)考虑低促性腺激素性性腺功能减退,病变定位于下丘脑或垂体。无颅内器质性或占位性病变,且病程中无炎症、外伤等继发因素,诊断特发性低促性腺激素性性腺功能减退症;戈那瑞林兴奋试验反应良好,提示病变在下丘脑。
通过性激素、垂体相关激素测定,鞍区MRI及常规生化检查等,容易与高促性腺激素性性腺功能减退症,因垂体前叶发育不良、垂体柄中断综合征、鞍区肿瘤等病变导致的垂体前叶多种激素分泌不足,以及慢性系统性疾病引起的青春期发育延迟相鉴别。
体质性青春期延迟和IHH在临床上通常较难鉴别,尤其在14~18岁的男性患者。根据2015年中华医学会内分泌学分会性腺学组制定的《特发性低促性腺激素性性腺功能减退症诊治专家共识》,为了提高临床操作的简便性,可以参考以下几方面进行综合判断[1]:
1.睾丸体积
隐睾或睾丸体积1~3ml,常提示IHH;体积≥4ml,提示青春发育延迟或部分性IHH。
2.基础状态LH水平
LH在0~0.7IU/L,提示IHH;LH≥0.7IU/L,提示青春发育延迟或部分性IHH。
3.戈那瑞林兴奋试验
静脉注射戈那瑞林100μg,0min和60min时测定LH水平。典型IHH患者LH峰值<8IU/L,如60min LH≥8IU/L,提示下丘脑-垂体-性腺轴启动,可能为体质性青春发育延迟或部分性IHH。
4.骨龄
IHH患者或体质性青春发育延迟者,骨龄一般落后于生物学年龄2~3年。体质性青春发育延迟者,骨龄进展到12岁时就会自发启动青春发育;如骨龄>12岁仍无青春发育迹象,且LH、FSH和睾酮水平低下,可确诊IHH而非青春发育延迟。
但需要注意的是,青春发育是一个连续变化的动态过程,临床医生不能仅依据某一个数据而忽视其他临床表现。因此IHH的诊断需综合考虑年龄、第二性征、性腺体积、激素水平和骨龄等诸多因素[2]。14岁尚无青春发育的男性,应进行青春发育相关检查。对暂时难以确诊者,可给予间断性小剂量性激素(如十一酸睾酮口服40mg/d)治疗,促进第二性征发育、身高增长及心理健康,定期评估睾丸体积和促性腺激素水平,观察是否有青春发育启动;随访观察应持续到18岁,以明确最终诊断。
而该患者本次就诊已24岁,骨龄17岁,第二性征发育迟缓,睾酮和促性腺激素水平均低下,而GnRH兴奋试验反应良好,IHH诊断明确,同时患者合并嗅觉障碍,故诊断为Kallmann综合征。
(二)IHH的治疗及随访
男性IHH治疗方案主要有3种,包括睾酮替代、促性腺激素和脉冲式GnRH生精治疗。可根据患者下丘脑-垂体-性腺轴的功能状态以及患者的年龄、生活状态和需求在3种方案中进行选择,并可互相切换。
确诊IHH后若患者暂无生育需求,睾酮替代治疗可促进男性化表现,小剂量开始逐渐增加至成人剂量。有生育要求的IHH患者则适合进行HCG/HMG联合生精治疗或脉冲式GnRH(GnRH泵)生精治疗。HCG/HMG联合生精治疗的常用方案为先肌内注射HCG 2000IU,每周2次,共3个月,其间调整HCG剂量,使血睾酮维持在10.41~17.35nmol/L(300~500ng/dl);然后联合肌内注射HMG 75~150IU,每周2~3次。既往研究报道,国内HCG/HMG联合生精治疗的有效性约为70%。初始睾丸体积和治疗过程中睾丸体积增大的幅度是预测精子生成的最重要指标。睾丸初始体积大于4ml是生精治疗成功的有利因素。GnRH泵生精治疗的常用起始剂量为GnRH(戈那瑞林)10μg/90min,带泵3天后如血LH≥1IU/L,则提示初步治疗有效。此后,每月随访1次,监测FSH、LH、睾酮和精液常规,调整戈那瑞林的剂量和频率,尽可能将睾酮维持在正常中值水平,稳定后可每3个月随访1次,依据患者的具体情况调整剂量。既往研究显示非隐睾患者2年精子生成率为100%。国内的治疗经验也提示,GnRH泵生精疗效优于HCG/HMG治疗[3,4]。
该患者目前24岁,有生育需求,且初始睾丸体积小,戈那瑞林延长试验中LH、FSH对GnRH反应良好,选择GnRH泵治疗可提高精子生成。GnRH泵治疗后随访第二性征发育情况、LH、FSH及睾酮,提示GnRH泵效果良好。进一步随访观察性激素可维持在正常水平,精液检查提示生精治疗有效,虽然精子活力欠佳,精子数目已达到正常人标准,存在受孕机会。该患者目前未婚未育,生育功能有待长期随访观察。
(三)IHH病因及遗传学研究进展
IHH可以散发或遗传,目前已经明确20多种基因突变可导致IHH[5],如kal1、fgfr1、fgf8、gnrh、gnrhr、prok2、prokr2、tac3、tacr3、dax1、nelf、chd7、sema3a、sox2、fezf1等;有5%的患者存在双基因或多基因突变;但仅1/3~1/2的患者可获得明确的基因诊断。
遗传的方式有多种:
1.X连锁隐性遗传,如经典的kal1基因突变,人群中男性患者远远多于女性。如与完全正常的女性生育后代,则男性患者的男性子代正常,而女儿则为携带者。
2.常染色体显性遗传,如fgfr1、fgf8,下一代子女均有可能发病,如父母中一方为杂合突变,则子代发病的可能性一般为1/2;如父母一方为纯合突变,则子代发病的可能性为100%。
3.常染色体隐性遗传,如prok2、prokr2,如父母一方为携带者,则下一代一般不会发病,但为携带者。
4.对于多基因突变者是否会遗传给下一代很难预测。要回答该患者是否会遗传的问题,需先行IHH相关基因筛查。如能明确病变基因可有助于了解其遗传模式;如果不能明确,则无法作出明确的答复。
【专家点评】
此病例为典型的Kallmann综合征,选用GnRH泵治疗效果良好。患者有生育需求,为促进优生优育,需动员患者进一步行基因检测以明确诊断,了解其遗传模式。
参考文献
[1]中华医学会内分泌学分会性腺学组.特发性低促性腺激素性性腺功能减退症诊治专家共识.中华内科杂志,2015,54(8):739-744.
[2]Melmed S,Polonsky KS,Larsen PR,et al. Williams Textbook of Endocrinology. 13th ed. Philadelphia:Elsevier/Saunders,2016:1075-1218.
[3]Gong C,Liu Y,Qin M,et al. Pulsatile GnRH is superior to hCG in therapeutic eff i cacy in adolescent boys with hypogonadotropic hypogonadodism. J Clin Endocrinol Metab,2015,100(7):2793-2799.
[4]黄炳昆,茅江峰,伍学焱,等.GnRH脉冲输注与HCG/HMG联合肌注对男性IHH患者生精治疗效果比较.中华医学杂志,2015,95(20):1568-1571.
[5]Boehm U,Bouloux PM,Dattani MT,et al. Expert consensus document:European Consensus Statement on congenital hypogonadotropic hypogonadism-pathogenesis,diagnosis and treatment. Nat Rev Endocrinol,2015,11(9):547-564.
来源:《复旦大学附属华山医院垂体疑难病多学科诊治病例精选》
作者:叶红英 王镛斐
页码:324-329
出版:人民卫生出版社
- 评价此内容

 3我要打分
3我要打分




 会员登录
会员登录
近期推荐
- 25岁女性无明显诱因体重进行性…
- 48岁绝经女性体检意外发现双侧…
- 21岁男性近半年体重下降约7kg …
- 57岁男子发热、呕吐、腹泻 不…
- 中年男性因外伤行颅脑手术后出…
- 24岁男青年嗅觉障碍伴第二性征…
- 55岁女性库欣病手术后缓解 出…
- 60岁女性2年间体重降低27kg且…
- 79岁女性既往体健 谁是高钙血…
- 45岁女性疲劳乏力精神萎靡 6个…

热点文章
- 还没有任何项目!
热门关键词
最新会议
- 2013循证医学和实效研究方法学研讨会
- 欧洲心脏病学会年会
- 世界帕金森病和相关疾病2013年会议
- 英国介入放射学学会2013年第25届年会
- 美国血液学会2013年年会
- 美国癫痫学会2013年第67届年会
- 肥胖学会 2013年年会
- 2013年第9届欧洲抗体会议
- 国际精神病学协会 2013年会议
- 妇科肿瘤2013年第18届大会
- 国际创伤压力研究学会2013年第29届…
- 2013年第4届亚太地区骨质疏松症会议
- 皮肤病协会国际2013年会议
- 世界糖尿病2013年大会
- 2013年国际成瘾性药年会
- 彭晓霞---诊断试验的Meta分析
- 武姗姗---累积Meta分析和TSA分析
- 孙凤---Network Meta分析
- 杨智荣---Cochrane综述实战经验分享
- 杨祖耀---疾病频率资料的Meta分析
友情链接
合作伙伴
Copyright g-medon.com All Rights Reserved 环球医学资讯 未经授权请勿转载!
网络实名:环球医学:京ICP备08004413号-2
关于我们|
我们的服务|版权及责任声明|联系我们
互联网药品信息服务资格证书(京)-经营性-2017-0027
互联网医疗保健信息服务复核同意书 京卫计网审[2015]第0344号
