10岁儿童肝功异常但无病毒感染 需肝移植才能救命
7岁儿童,因上呼吸道感染查体,偶然发现血小板减少,脾大。当地医院进一步检查显示,肝功能异常,但甲、乙、丙、丁、戊型肝炎病毒学指标皆为阴性,合并有食管静脉曲张和出血性胃炎。10岁时再次入院,几经周折,医生终于确诊,竟是患有一种罕见的遗传性疾病。那么,该患儿患有何种疾病,有无治愈的可能?
患者,男,10岁,内蒙古人。主因“发现脾大、肝功能异常3年”入院。
入院情况:患儿3年前因上呼吸道感染查体时发现血小板减少,脾大,就诊于当地医院,化验:血小板减少,69×109/L,肝功能异常,ALT 70.38U/L,AST 71.1U/L,ALP 318.51U/L,GGT 57.48U/L,Alb 31.4g/L,TBIL 27.45μmol/L,DBIL 10.46μmol/L。甲、乙、丙、丁、戊型肝炎病毒学指标阴性。B超示:肝弥漫性改变,脾大,上下腔静脉内径较同龄明显变细,右心增大。头颅MRI:左侧颞叶前下方蛛网膜囊肿。骨髓检查未见明显病理改变,嗜血性组织细胞增多。眼科未发现K-F环。胃镜示:食管静脉曲张,糜烂、出血性胃炎,十二指肠球炎。诊断为:①肝硬化,脾功能亢进,食管静脉曲张;②糜烂出血性胃炎;③十二指肠球炎。给予保肝、保护胃黏膜治疗,并间断入院复查,患儿饮食、一般情况好,无腹胀、恶心、呕吐、纳差、乏力不适,不伴发热、咳嗽、胸闷、心悸、气短,无腹痛、腹泻,无下肢水肿,大便正常,近1年小便间断黄染。B超1年前提示少量腹水。半年前复查PLT 39×109/L,ALT 42.9U/L,AST 56.4U/L,GGT 48.4U/L,TBIL 74.6μmol/L,DBIL 15.7μmol/L,IBIL 58.8μmol/L。3天前来我院门诊眼科查K-F环阴性,北京朝阳医院查24小时尿铜结果待回报,为进一步诊治今收入院。既往幼时易患上呼吸道感染。个人史和家族史无特殊。查体:体型偏瘦,皮肤、巩膜轻度黄染,全身浅表淋巴结未触及肿大。心肺没有异常体征。腹饱满,腹壁静脉显露,肝肋下未及,脾肋下5厘米可以触及,质中等,无触痛,移动性浊音阳性。
初步诊断:患儿有如下特点:①患儿,发育不良,有慢性肝功能受损和门脉高压表现;②既往多次查各种嗜肝病毒标志物阴性;③没有明确的肝病家族史。这种慢性肝病往往不会是病毒性的,常常与先天或遗传性肝病相关,如铜代谢异常的Wilson病,α1-抗胰蛋白酶缺乏症,先天性肝纤维化等等,后天形成的如布-加综合征,肝小静脉阻塞症和门脉海绵样变性,无论是哪种情况,都需要进一步的检查,目前有明确门脉高压,脾功能亢进,食管静脉曲张,可以考虑肝硬化原因待查,具体的原因将在完善一些代谢指标的检查和影像学检查后来确定。目前治疗以对症治疗为主,待诊断明确后进一步确定治疗方案。
鉴别诊断:①肝豆状核变性:为常染色体隐性遗传病,因铜代谢异常导致其在各种器官特别是肝脏和脑中过量积聚,临床可表现为肝脏和神经系统功能障碍,并可有K-F环、甲床变蓝等典型改变,实验室检查可见血清铜蓝蛋白降低,24小时尿铜升高,头颅磁共振检查可见脑室扩大、普遍萎缩和基底节损害等,必要时可行肝活检和铜定量测定以明确诊断。但本患儿智力正常,无神经系统功能障碍,眼科未见K-F环,铜蓝蛋白无明显减低,可以进一步查24小时尿铜。②病毒性肝炎所致的肝硬化:患儿年纪小,乙、丙型肝炎标记物均阴性,可除外乙、丙型肝炎肝硬化。需除外巨细胞病毒、EB病毒感染所致肝损伤。③先天性肝纤维化:是一组少见的常染色体隐性遗传性疾病,临床以门脉高压和肝功能正常为特点,多合并常染色体隐性遗传性多囊肾和(或)肝内外胆管发育异常。肝脏病理示汇管区显著增宽,纤维组织堆积,小胆管异常增生,可出现不同程度扩张,肝小叶结构完整。④布-加综合征:该病属肝后型门脉高压症,系各种病因造成的肝静脉回流受阻肝窦压力升高导致门脉高压,临床上可表现为肝脾大、腹水、食管胃底静脉曲张等,后期可形成肝硬化。⑤特发性门脉高压症:为肝内门静脉分支发生闭塞性纤维化及硬化,病理形态上有肝内门静脉分支带状纤维化和狭窄,窦状隙有胶原纤维沉着。发病机制不清,可致窦前性门脉高压而出现静脉曲张出血及脾脏肿大。肝功能一般正常,B超、CT可无明显异常。⑥患者入院后查非结合胆红素较高,查网织红细胞计数、外周血细胞形态、Coombs试验、Ham试验除外溶血。注意患儿腹部CT回报,了解门脉成像情况。必要时肝穿病理检查明确诊断。
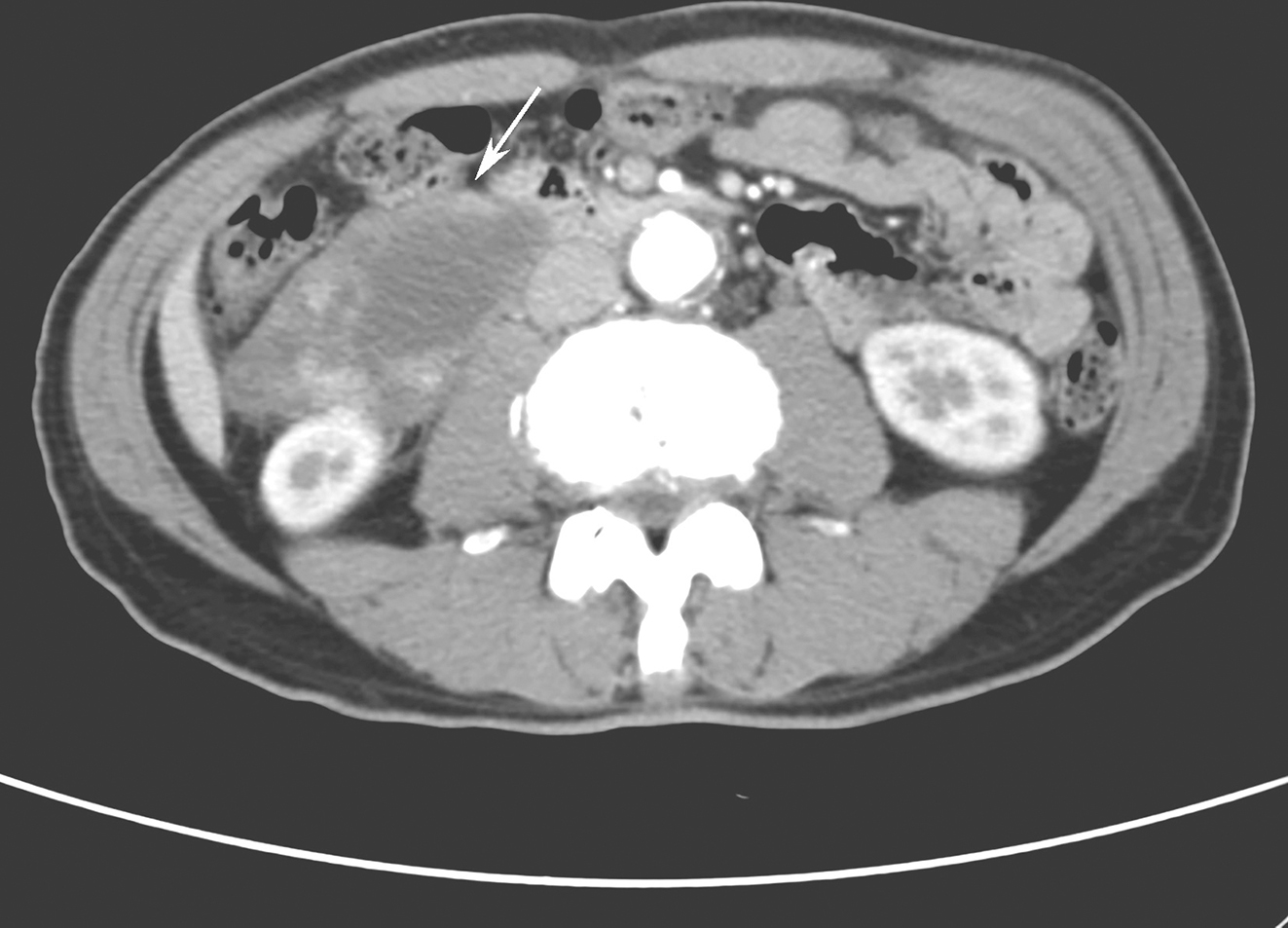
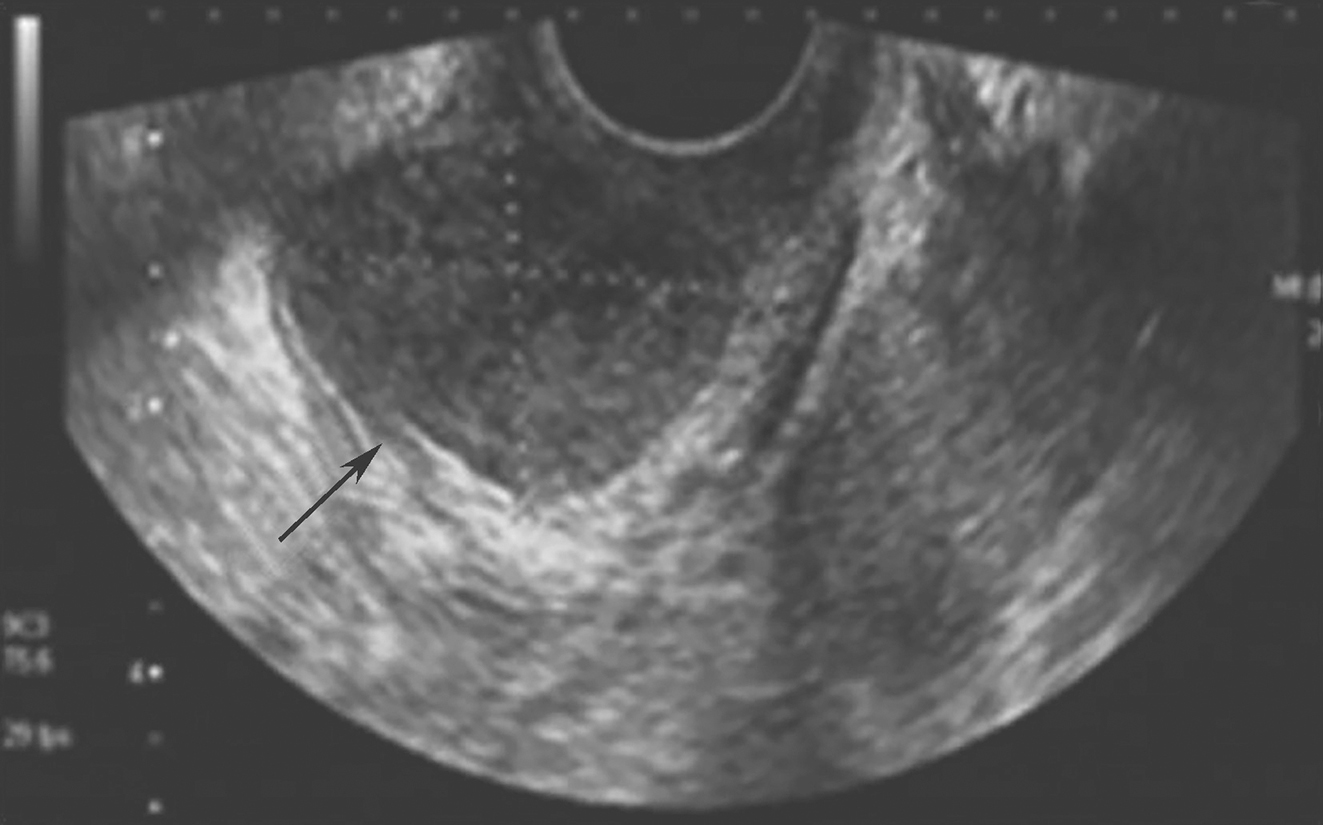
诊疗经过:患儿入院后一般情况好,由于北京朝阳医院查24小时尿铜38.7μg,尿铜不高,复查铜蓝蛋白正常,没有K-F环表现,尽管以上某些症状和体征可能在罕见病例中出现阴性,但全部阴性的患者诊断这种疾病仍然牵强,入院后的普通化验没有更多的阳性发现,重复确认了患儿以前的阳性化验: WBC 3.78×109/L,PLT 53×109/L,WBC、PLT减低,肝功能异常: ALT 29U/L,AST 42U/L,ALP 397U/L,Alb 31.1g/L,TBIL 60.76μmol/L,PT 16.3秒,病原学均为阴性。血清铁9.1μmol/L,总铁结合力47.2μmol/L。血清铁正常,除外血色病。病原学检查均为阴性,如行病理检查应该会提示或除外先天性肝纤维化或特发性肝纤维化,但由于患者PT轻度延长和腹水存在,因此上级医师认为在患者病情稍稳定的时候再行肝穿刺活检。在入院第4天腹部B超显示:①肝右叶可见多发高回声结节样病变,性质待定;②弥漫性肝病,门脉海绵样变,脾大,脾静脉增宽;③门脉主干血流速度正常;④腔静脉血流通畅。腹部CT示:①肝脏多发类圆形强化灶,肝硬化再生结节可能性大;②肝硬化、脾大、门脉高压、侧支循环形成。提示患儿有可能是肿瘤或有弥漫的肝硬化结节,CT门脉成像提示肝门区门静脉正常结构消失。胃镜显示:食管胃底静脉曲张(重度),慢性浅表性胃炎,十二指肠球炎。腹部MRI平扫+增强显示:①肝硬化,脾大,食管胃底、脾静脉迂曲扩张,少量腹水。②门静脉未见显示,门静脉缺失或闭塞可能大,考虑先天性异常。③肝脏多发类圆形异常信号病灶,肝硬化不典型增生结节可能,建议观察或进一步检查。肠系膜血管静脉造影检查:脾脏体积增大,门静脉系统均未显影,回流血液入胃底-食管静脉及腰深静脉,未见脾肾静脉分流。下腔静脉通畅。印象:门静脉闭塞。
新的临床证据提示了先天性门脉缺失的可能。先天性门静脉缺失是指血液经肠系膜上静脉及脾静脉经肝的侧支回流到体循环静脉,而没有门静脉。首次是在1793年被发现,有学者随后提出将门静脉分流分为2种不同的类型:Ⅰ型为门静脉所有的血流经肝脏的侧支循环回流到体循环,这种类型的分流叫完全分流;Ⅱ型为门静脉的部分血液直接经肝脏回流,这种类型的分流叫部分分流。Ⅰ型患者一旦侧支被阻塞,患者的结局将不乐观,因为这是门静脉回流的唯一途径,然而Ⅱ型患者能经血管结扎成功治疗。如果是先天的缺失,这名患儿很有可能是Ⅰ型患者,还有需要指出的是,B超显然在诊断这种疾病上敏感性下降,其观察到的门脉往往是开放的侧支循环的血流,而进行门脉成像会观察到实际的血管走行,而肝脏多发类圆形强化灶,考虑门静脉缺失,动脉灌注异常所致。由于高度怀疑患儿肝脏是动脉供血,因此,需要取消肝穿检查。
患者家庭无经济条件行肝移植术,建议其转外科行肠-腔静脉分流术,以缓解食管胃底静脉曲张,预防大出血危及生命。转入外科后发现肺动脉高压严重,手术风险高,患儿出院,后失访。
最终诊断:先天性门脉缺失。
文献复习:先天性门脉缺失最早在1961年被报道,随后不断有病例报道,此疾病可以引起一系列的临床表现,简单可以分为两类,一类是先天性的异常,包括肝脏和心脏的病变,这可以解释这种患者常常会伴发心脏的疾病,此患者也在术前发现肺动脉高压表现,这种情况也见于文献报道,从而丧失了手术的机会,一类是继发的病变,包括肝脏由于血供不足引起的发育不良,肝功能异常,并引起局灶性结节性增生(focal nodular hyperplasia,FNH)和肝脏肿瘤,有些患者会出现结节性再生性增生(nodular regenerative hyperplasia of the liver,NRH),此患者也有这种表现,在影像学上提示肝脏结节表现,原因可能与动脉供血相关,但具体是FNH、NRH还是肝脏肿瘤,由于缺乏病理难以确定其为良性还是恶性,但据报道大多数这种结节还是良性的,患者的肝脏可大可小,如果肝脏增大则是肝内结节增生的结果,如果肝脏减小是由于肝脏血供不足引发,此患儿肝脏没有明显增大,在一定程度上支持其良性结节病变的可能。
此疾病由于与胚胎发育时候的血管发育异常有关,因此最常见的并发症是心血管疾病,包括充血性心衰和三尖瓣反流,但这个患儿并没有发现这些常见的疾病,但肺动脉高压存在,这种不伴随心脏明显病变的情况也常见于这类患者,其次的病变还包括:先天性的胆道闭锁,胆总管囊肿,肝内胆囊,肾脏囊性发育不良,肾脏双侧输尿管梗阻,膀胱输尿管逆流,尿道下裂,溃疡性结肠炎,肠息肉,腹股沟疝,右上颌骨发育不良,骨发育异常和轻度畸形,五指短小等等。但此患儿除了发育迟缓外没有发现上述的并发症,由此看来,这种疾病的表现可以是多种多样的损伤,可能与患者先天发育的受损范围有关。
患者可以合并各种程度的肝脏疾病或者肝功能正常,其肝脏的损伤程度主要因为门脉供血的缺失和肝脏结节的产生,由于门脉高压和肝脏对血糖、血氨的影响,临床可以见到肝肺综合征、肝肾综合征、肝性脑病,此患儿有轻度的腹水和肝脏功能受损,而且合并肺动脉高压(这种表现在最近的文献中也有报道),这种情况可能属于病例报道中最常见的临床表现。
对此种患者的治疗依赖于患者的临床、生化以及影像学改变,如果患者没有临床症状,则可以先不需治疗,对于那些肝脏有结节的患者,要考虑其大小和组织学的改变,如果患者新生物迅速变大,则发生恶性的可能性大,就可以选择肿物切除或者肝脏移植,如果组织学提示是恶性的也应该进行进一步治疗,切除结节的手术一般并不复杂,而且术后恢复时间并不比其他疾病患者慢,可能动脉供血的血流足够维持患者的肝脏再生,目前来讲,越来越多的外科医师认为这种患者的肝移植指征是内科无法控制的患者,如胆道闭锁引起的肝硬化,肝脏弥漫的肝母细胞瘤,严重的肝性脑病,如果患者有肺部并发症也可以进行肝移植治疗,因为如果肺部血管压力过高是肝移植的禁忌证,肝移植之后一般患者恢复良好,除了肝脏的功能恢复以外,此疾病引起的其他并发症也会得到缓解。
(李海)
参考文献
1. Yancy Ag,Jeffries Cp,Miller Rm.Congenital absence of the portal vein.JNatlMed Assoc,1961,53: 119-121.
2. Tannuri U,Galvao F,Leal AJ,et al.Congenital absence of the portal vein: a complex disease with multiple manifestations and types of treatment.Eur JPediatr Surg,2011,21(4): 269-272.
3. Hino T,Hayashida A,Okahashi N,et al.Portopulmonary hypertension associated with congenital absence of the portal vein treated with bosentan.Intern Med,2009,48(8): 597-600.
4. Asran MK,Loyer EM,Kaur H,etal.Case 177: Congenitalabsence of the portal vein with hepatic adenomatosis.Radiology,2012,262(1): 364-367.
5. Chandler TM,Heran MK,Chang SD,et al.Multiple focal nodular hyperplasia lesions of the liver associated with congenital absence of the portal vein.Magn Reson Imaging,2011,29(6): 881-886.
6. Scheuermann U,Foltys D,Otto G.Focal nodular hyperplasia precedes hepatocellular carcinoma in an adult with congenital absence of the portal vein.Transpl Int,2012,25(5): e67-68.
7. Uchida H,Sakamoto S,Shigeta T,et al.Living donor liver transplantation with renoportal anastomosis for a patientwith congenital absence of the portal vein.Case Rep Surg,2012,2012: 670289.
8. Kasahara M,Nakagawa A,Sakamoto S,et al.Living donor liver transplantation for congenital absence of the portal vein with situs inversus.Liver Transpl,2009,15(11): 1641-1643.
来源:《消化科病例分析:入门与提高》
作者:辛永宁 柏愚
页码:302-305
出版:人民卫生出版社
- 评价此内容

 3我要打分
3我要打分


 会员登录
会员登录
近期推荐
- 48岁肥胖男性反酸、胸骨后烧灼…
- 39岁男子腹痛伴便秘2年 别不当…
- 青年女性反复上腹痛伴胃灼热感…
- 58岁女性转移性右下腹疼痛未重…
- 一大叔右上腹隐痛消炎治疗无效…
- 海鲜和烧烤下肚 年轻男子腹痛…
- 42岁男子干农活时呕血两次 问…
- 48岁女性反复右上腹隐痛1年多 …
- 45岁女性上腹痛、间断双下肢水…
- 45岁男子受凉后乏力 自服感冒…

热点文章
- 还没有任何项目!
热门关键词
最新会议
- 2013循证医学和实效研究方法学研讨会
- 欧洲心脏病学会年会
- 世界帕金森病和相关疾病2013年会议
- 英国介入放射学学会2013年第25届年会
- 美国血液学会2013年年会
- 美国癫痫学会2013年第67届年会
- 肥胖学会 2013年年会
- 2013年第9届欧洲抗体会议
- 国际精神病学协会 2013年会议
- 妇科肿瘤2013年第18届大会
- 国际创伤压力研究学会2013年第29届…
- 2013年第4届亚太地区骨质疏松症会议
- 皮肤病协会国际2013年会议
- 世界糖尿病2013年大会
- 2013年国际成瘾性药年会
- 彭晓霞---诊断试验的Meta分析
- 武姗姗---累积Meta分析和TSA分析
- 孙凤---Network Meta分析
- 杨智荣---Cochrane综述实战经验分享
- 杨祖耀---疾病频率资料的Meta分析
友情链接
合作伙伴
Copyright g-medon.com All Rights Reserved 环球医学资讯 未经授权请勿转载!
网络实名:环球医学:京ICP备08004413号-2
关于我们|
我们的服务|版权及责任声明|联系我们
互联网药品信息服务资格证书(京)-经营性-2017-0027
互联网医疗保健信息服务复核同意书 京卫计网审[2015]第0344号
