上腹痛和指(趾)甲增厚 一种罕见的致命性疾病
60岁男性,1个月前无明显诱因出现上腹部疼痛,与体位、饮食和呼吸无明显关系。医生怀疑为消化性溃疡。然而,医生注意到,患者还有一个典型的特征,四肢指(趾)甲增厚,萎缩,变脆。经过两次胃肠镜和病理检查证实,却是一种非常罕见的疾病。
患者,男,60岁。主诉:上腹痛1个月余。
入院情况:患者于1个月前无明显诱因出现上腹部疼痛,呈阵发性胀痛和隐痛不适。每次持续约30分钟至数小时,尚能耐受。疼痛无放射,与体位、饮食和呼吸无明显关系。自服“陈香露白露片”后症状可减轻,但反复发作。病程中无恶心、呕吐、反酸、嗳气、呕血、黑便;无腹胀、便秘、肛门停止排便排气;无发热、黄疸;无关节痛、皮疹、精神异常。今来我院就诊,门诊以“腹痛原因待查”收住我科。发病以来,患者精神睡眠差,食欲略有下降,大便稀,无血便及黏液血便,小便正常,体重下降4kg。既往有慢性腹泻病史1年,未明确诊断。有高血压病史5年余,最高血压130/100mmhg,平时服用“氨氯地平片、美托洛尔缓释片”治疗,血压控制可。有“血糖升高”史,糖尿病未确诊。否认肝炎、结核等传染病史,否认外伤、手术及输血史,未发现药物过敏史。无酗酒、吸烟、吸毒等不良嗜好。否认到过传染病、地方病流行地区。否认有工业毒物、粉尘、放射性物质接触史。30岁结婚,配偶及子女身体健康。父母已故(死因具体不详)。家族成员中无家族性恶性肿瘤家族史,否认有家族性遗传性疾病。入院体格检查:T 36.2℃,P 7次/分,R 18次/分,BP 118/80mmHg。发育正常,营养中等,神志清楚。慢性病容,面色黝黑,头发稀疏,躯干四肢多处皮肤片状色素沉着,四肢末端皮肤发黑。全身表浅淋巴结未及肿大。巩膜无黄染。气管居中。甲状腺无肿大。双肺呼吸音清,未闻及干湿性啰音。HR 70次/分,律齐,心脏各瓣膜区未闻及病理性杂音。腹稍膨隆,全腹软,无压痛,无反跳痛,未触及肿块,肝脾肋下未及,Murphy征阴性。双肾区无叩击痛。移动性浊音阴性。肠鸣音4次/分。双下肢无水肿,四肢指(趾)甲增厚,萎缩,变脆。生理反射存在,病理反射未引出。实验室检查:血细胞分析:Hb 173g/L,PLT 206×109/L,WBC 8.05×109/L,中性粒细胞65.6%,淋巴细胞25.5%。肾功能及电解质:BUN 3.6mmol/L,CR 89.0μmol/L,K+ 3.93mmol/L,Na+ 143.3mmol/L。血淀粉酶53U/L,血脂肪酶273U/L。肝功能:TP 53.9g/L,Alb 33.3g/L,ALT 17U/L,AST 14U/L,ALP 46U/L,GGT 14U/L,TBIL 14.6μmol/L,DBIL 12.2μmol/L。凝血功能、肿瘤标志物、尿常规均未见明显异常;病毒标志物示HBsAb阳性,余无异常;大便常规示:黄色半稀便,未检出寄生虫卵,潜血阴性。
初步诊断:①上腹痛原因待查:消化性溃疡可能?②高血压2级。在上腹痛的病因中,消化性溃疡比较多见,患者症状不典型,服用制酸药后症状可减轻,故第一诊断考虑消化性溃疡。其余可能的诊断:①慢性胃炎:主要表现为上腹痛或不适、上腹胀、早饱、嗳气、恶心等消化不良症状。该患者本病不能排外,需进一步行胃镜检查明确。②胃癌:该患者有上腹痛,体重下降,但其他表现不支持。③功能性胃肠病(FD,IBS):功能性消化不良的主要症状是上腹部疼痛、上腹部烧灼感、餐后饱胀或早饱。肠易激综合征是一组包括腹痛、腹胀、排便习惯和大便性状异常的症状群。该患者有一年以上慢性腹泻,虽无贫血、黑便等报警症状,但该患者近期有体重下降,且未行胃肠镜检查排外器质性疾病,因此该诊断留到最后考虑。
鉴别诊断:患者为慢性腹痛,故需和引起慢性腹痛的疾病鉴别。①腹腔脏器的慢性炎症如反流性食管炎、慢性胆囊炎、胆结石及胆道感染、慢性胰腺炎、结核性腹膜炎、溃疡性结肠炎、克罗恩病等。该患者均缺乏相应的表现和证据,可能性小。②腹腔器官肿瘤如结肠癌、胰腺癌、肝癌等。该患者有体重下降,但无贫血、便血、黄疸等肿瘤表现,肿瘤标志物未见异常,需进一步完善胃肠镜、腹部影像学检查排外。③中毒与代谢障碍如铅中毒、尿毒症等基本排除。
该患者目前主要诊断为:上腹痛原因待查:消化性溃疡可能?因此下一步首先安排胃镜检查。为排外胆道疾病和肿瘤,拟行腹部B超、CT检查。因该患者有躯干四肢多处皮肤片状色素沉着,四肢末端皮肤发黑表现,需考虑是否存在肾上腺皮质功能减退,故拟行促肾上腺皮质激素和皮质醇测定。患有高血压病史5年,未进行系统检查,故安排了心脏超声和动态血压检查。患者有1年以上慢性腹泻,未明确诊断,因而需行肠镜检查辅助诊断。其四肢指(趾)甲增厚,萎缩,变脆,需考虑甲癣可能,故请皮肤科会诊行真菌检查。入院后给予泮托拉唑抑制胃酸、铝镁加混悬液保护胃黏膜及对症治疗。
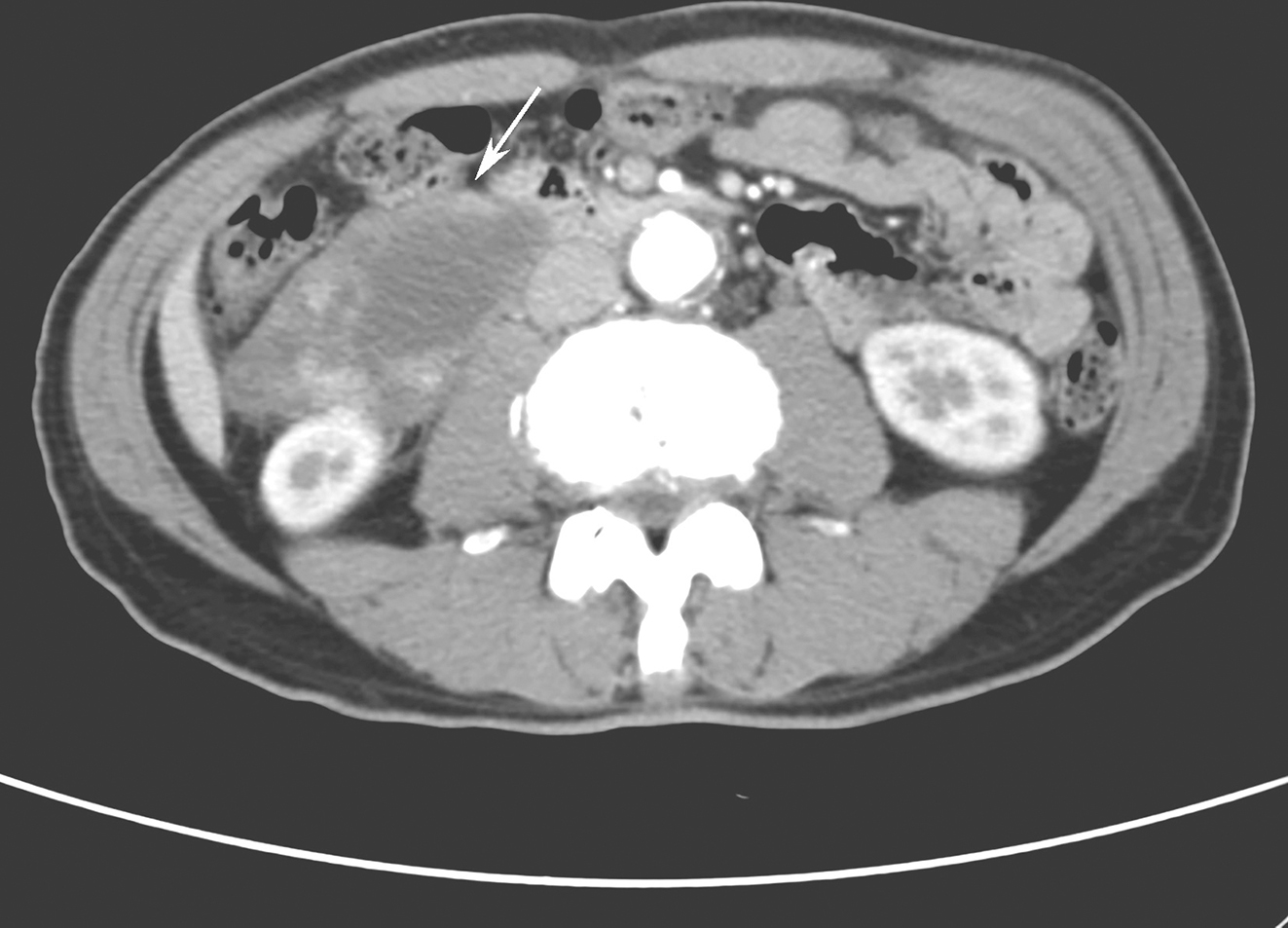
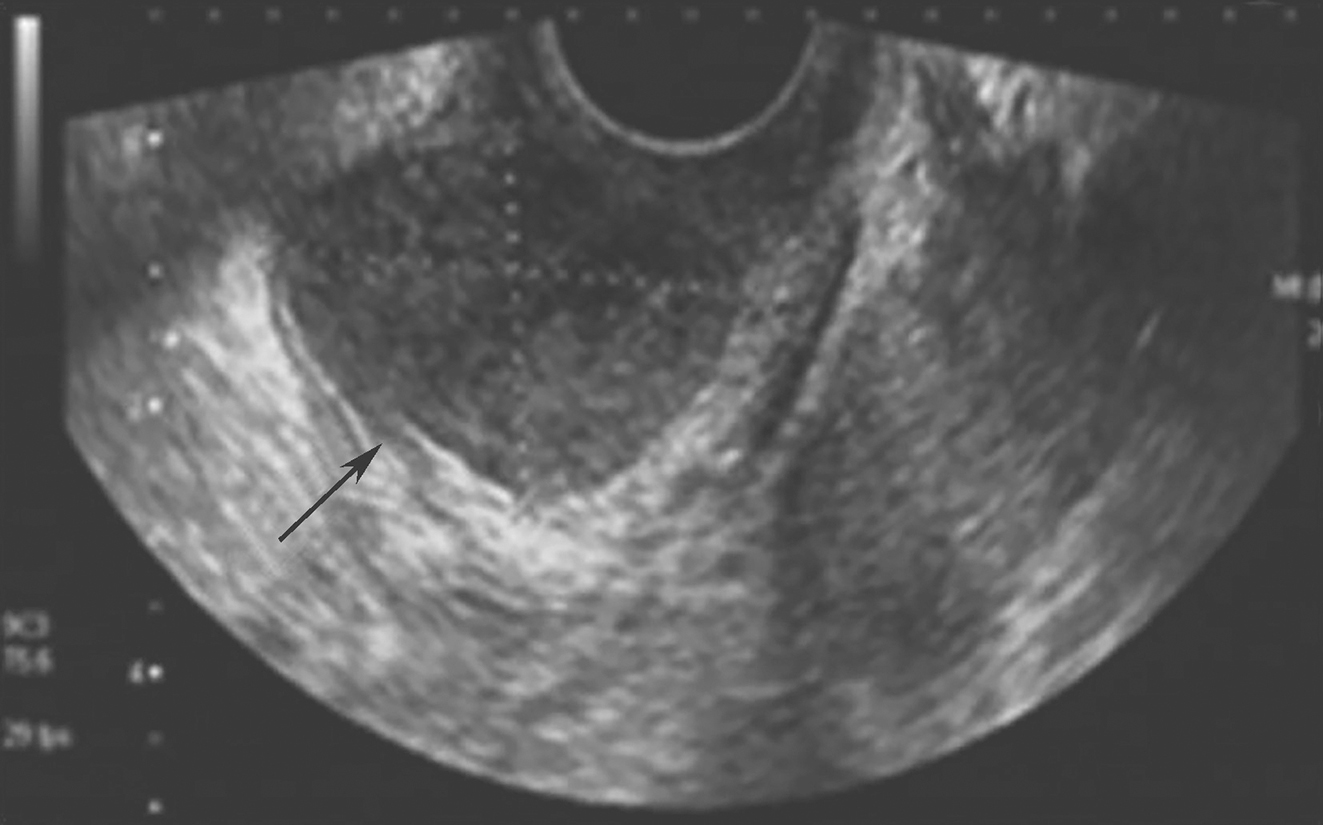
诊疗经过:心电图:窦性心律。胸平片示:心脏、大血管呈“主动脉型”改变,左心室轻度增大。腹部B超:肝多发囊肿,胆囊炎,胆囊结石,胆囊多发息肉。腹部CT示:肝脏多发低密度病变,考虑为肝囊肿;胆囊炎,胆囊结石。心脏彩超:左心房、右心房增大,右心室稍增大;主动脉内径稍增宽;室间隔增厚;左室舒张功能减退;二、三尖瓣少量反流。动态血压:昼夜血压呈非杓型,全天血压偶见升高。促肾上腺皮质激素3.33pmol/L,皮质醇497.2nmol/L,均在正常范围。免疫全套:IgM 0.37g/L,CD3 49.5%,CD8 12.1%,ENA抗体谱阴性。胃镜:胃底黏膜皱襞粗大,充血水肿,见弥漫性出血点,息肉样隆起,表面充血糜烂。胃窦黏膜皱襞充血水肿,黏膜凸凹不平,息肉样隆起,表面充血糜烂。十二指肠球部大弯侧前壁见大小约0.8cm×1.2cm溃疡,表覆白苔,黏膜充血水肿,球后见弥漫性大小不等息肉样隆起。病理:(胃窦部)黏膜组织慢性炎伴急性炎。送检胃黏膜固有层内黏膜腺体结构存在,大小不一。间质充血水肿,伴淋巴细胞、浆细胞及嗜中性粒细胞浸润。13 C呼气试验阳性。结肠镜:肛门见环状内痔,近乙状结肠处起见0.2cm×0.3cm扁平结节,表面光滑。乙状结肠、降结肠、横结肠、升结肠黏膜充血水肿,血管网模糊,见多发散在亚蒂或无蒂息肉,大小约0.2cm×0.2cm~0.4cm×0.5cm,表面光滑。回盲部见回盲瓣变形,形成3.0cm×3.5cm隆起,表面分叶,充血水肿,糜烂。对侧肠壁见1.2cm×1.2cm息肉样病变。回肠末段见散在扁平结节,大小约0.2cm×0.2cm,表面光滑。病理:(回盲部、横结肠、降结肠)黏膜息肉样增生。皮肤科会诊:甲屑镜检阳性。
从胃镜检查结果分析,符合初步诊断消化性溃疡的判断,患者的上腹痛考虑为十二指肠溃疡所致,且患者入院后经抑制胃酸分泌等治疗后疼痛缓解也证实了这一点。腹部B超显示胆囊炎,胆囊结石,胆囊多发息肉。鉴于患者既往从未有胆囊炎,也未出现右上腹部疼痛,Murphy征阴性。考虑为常见的无痛性胆囊结石,与此次腹痛关系不大,建议患者择期外科手术。
此次的胃肠镜检查结果发现的胃十二指肠和结肠的多发息肉却引出另外一个问题,患者慢性腹泻是否与此相关?多发息肉的诊断是什么?
经促肾上腺皮质激素和皮质醇测定,可排除肾上腺皮质功能减退症。那患者躯干四肢多处皮肤片状色素沉着,四肢末端皮肤发黑的原因是什么?
本病例临床主要特点为:①老年男性,病程1个月余,上腹部疼痛为阵发性胀痛和隐痛不适,服“陈香露白露片”后症状可减轻,但反复发作;②既往有慢性腹泻病史和高血压病史;③面色黝黑,头发稀疏,躯干四肢多处皮肤片状色素沉着,四肢末端皮肤发黑。四肢指(趾)甲增厚,萎缩,变脆;④胃肠道多发息肉。
为了明确诊断,我们需把临床上可见到的胃肠道多发息肉和皮肤色素沉着疾病纳入考虑。
1.黑斑息肉综合征 又称Peutz-Jeghers综合征(Peutz-Jeghers syndrome,PJS)。PJS是一种常染色体显性遗传病,多在青少年时期发病,60岁以上少见。主要特点是胃肠道多发性息肉和皮肤黏膜色素沉着。皮肤黏膜色素沉着以唇、口周、颊黏膜、手掌、足底、指(趾)、眶周围多见,边缘清楚,大小不等,形态不规则,不高于皮肤。息肉一般在100个以内,也可单发。息肉可分布于整个胃肠道,尤以小肠多见,其次为大肠、胃和十二指肠。病理上为错构瘤样息肉,虽然错构瘤被认为是一种良性病变,但黑斑息肉综合征具有发展成腺癌的倾向。主要临床症状为腹痛、腹胀、呕吐,部分患者可出现便血等表现。腹痛多由胃肠道多发息肉引起,可反复发作,甚至引起肠梗阻肠套叠。本例患者有多发息肉和色素沉着表现,但无家族遗传疾病史;色素沉着为躯干四肢多处皮肤,而非PJS的唇、口周等部位的色素沉着;且病理学也未发现错构瘤改变,故PJS的可能性小,为排除病理学上的误差,拟再次行胃肠镜及病理检查。
2.Gardner综合征 可发生在任何年龄,从2个月至70岁之间均可出现症状。病因未明,一般有明显家族史,男女均可发病。临床症状常为腹泻、黏液便或血便。肠镜检查结肠多发息肉,数量可达100个以上,均为腺瘤性息肉,可发生癌变。胃、十二指肠等消化道其他部位多发息肉亦多见。肠外表现为:①硬纤维瘤;②骨疣和骨瘤:主要见于头颅、上下颌、四肢长骨,均为良性;③牙齿异常;④其他软组织肿瘤:多为皮脂腺囊肿、纤维瘤表皮囊肿,脂肪瘤等。本病例有腹泻和胃肠多发息肉表现,但皮肤改变和病理均不支持Gardner综合征。
3.黑棘皮病 黑棘皮病是以皮肤色素增生、角化过度、疣状增殖为特征的少见皮肤病,分为良性和恶性。良性黑棘皮病可以是特发的、遗传的,或与内分泌疾病和药物有关,皮损范围小,有一定自限性,部分患者可自愈。恶性黑棘皮病皮损发展快,严重而广泛,色素沉着明显且不限于增厚皮损,黏膜及皮肤黏膜交界处亦有典型皮损,眼和唇周有乳头瘤样增生、指甲变脆有纵形嵴,且掌跖有特征性“天鹅绒样”改变,皮损处常有瘙痒或刺激症状,是恶性肿瘤伴发皮肤损害的一种表现形式,常具有其他内脏恶性肿瘤所具有的3种皮肤标志,即Leser Trelat征(急性发疹性脂溢性角化病皮肤表现)、自然红色皮肤乳头瘤病和掌跖角化过度。几乎100%伴发内脏癌,多数好发胃腺癌。本例患者皮肤改变非典型黑棘皮病,病理学未发现肿瘤证据,但仅仅一次病理学检查并不能完全排外。
4.Cronkhite-Canada综合征 即Cronkhite-Canada syndrome(简称CCS),又称胃肠道息肉-色素沉着-秃发-指(趾)甲萎缩综合征。其诊断的主要依据为成年发病,男性多见;无家族史;胃肠道广泛多发息肉;有外胚层病变如皮肤色素沉着、脱发、指(趾)甲萎缩等;临床表现为腹泻、腹痛、食欲不振、纳差、体重下降等症状。该患者具备以上特征,诊断为此病可能性大、但患者患有指甲真菌病、临床表现不典型、且还需进一步排外上述疾病。
其他胃肠道多发息肉疾病如Cowden综合征、幼年型息肉病、家族性结肠息肉病、多发肿瘤(Turcot)综合征虽有胃肠道多发息肉表现,但均有家族遗传性及其他特征,均可以排外。
为进一步排外黑斑息肉综合征和肿瘤,故给患者安排了第二次肠镜和病理检查,结果如下。
胃镜:食管表面粗糙,下段可见散在息肉样隆起,大小约0.2~0.3cm,表面光滑;胃底、胃体、胃角、胃窦黏膜粗糙,充血水肿,见弥漫性出血点及多发息肉样隆起,大小约0.2~1.0cm,表面光滑。十二指肠球部及降段黏膜粗糙,充血水肿,见弥漫性出血点及多发息肉样隆起,大小约0.2~0.5cm,表面光滑。病理:(胃窦部)复合增生性息肉,送检胃黏膜息肉样增生,间质内腺体大小不一,间质疏松水肿,血管扩张增生,弥漫性淋巴细胞、浆细胞及嗜中性粒细胞浸润。
结肠镜检查:肛门见环状内痔,近乙状结肠处起见0.2cm×0.3cm扁平结节,表面光滑。乙状结肠、降结肠、横结肠、升结肠黏膜充血水肿,血管网模糊,见多发散在亚蒂或无蒂息肉,大小约0.2cm×0.2cm~0.6cm×0.8cm,表面光滑。乙状结肠、降结肠病变稍轻。回盲部见回盲瓣变形,形成3.0cm×3.5cm隆起,表面分叶,充血水肿,糜烂。对侧肠壁见1.2cm×1.2cm息肉样病变。回盲部黏膜粗糙。回肠末段见散在扁平结节,大小约0.2cm×0.2cm,表面光滑。病理检查:(回盲部、升结肠、横结肠)黏膜息肉样增生伴慢性炎,组织学见腺体扩张,纤维间质增生,小血管增生。
经过两次胃肠镜和病理检查,结合临床表现,排外了黑斑息肉综合征,Gardner综合征和黑棘皮病。诊断为:Cronkhite-Canada综合征。可是仍有疑问,虽然指(趾)甲病变并非诊断Cronkhite-Canada综合征必需,但患者趾甲病变是否完全可以用指(趾)甲真菌病解释?因为患者存在四肢所有指(趾)甲增厚、萎缩、变脆。故我们请皮肤科再次在不同部位镜检,并培养,结果为阴性。患者入院后予泮托拉唑抑酸,铝镁加混悬液、谷氨酰胺胶囊保护胃肠道黏膜及对症、支持治疗后腹痛好转,腹泻改善。拟给小剂量皮质激素治疗,但患者担心激素副作用不愿口服激素,予营养支持、对症治疗后病情好转出院。
最终诊断:Cronkhite-Canada综合征。
文献复习:Cronkhite-Canada综合征(Cronkhite-Canada syndrome,CCS),国内又称胃肠道息肉-色素沉着-秃发-指(趾)甲萎缩综合征。是由Cronkhite-Canada于1955年首先报道的一组临床综合征,国内1985年首次报告本病。
Cronkhite-Canada综合征(CCS)是一种少见病,但它在世界范围内都有分布。1955—1985年间全世界共有154例报告,其中日本报告110例,占71.4%。到1997年报告数达到280例,1985—2006年我国有35例报道。CCS患者无息肉病家族史,是一散发的、非遗传性的疾病。发病年龄从24~86岁,平均60岁,80%以上的患者发病年龄超过50岁。CCS的病因和发病机制不明。与消化性溃疡以及溃疡性结肠炎相似,精神刺激、劳累、长期服药是常见的诱发因素。可能的发病机制有:代谢和内分泌失衡、免疫功能紊乱,感染,炎症、营养缺乏,中毒和先天异常等。
CCS起病相对较急,大多数患者从起病到确诊为3个月到1年不等。最常见的临床表现是腹泻、味觉减退、体重减轻、厌食、全身乏力以及脱发、甲萎缩或甲脱失、色素沉着、水肿、贫血和舌炎等,部分患者有口干和口腔感觉异常。最常见的首发表现是腹泻,或味觉减退。外胚层病变(脱发、甲萎缩和色素沉着)一般比其他临床表现晚几周至几个月,但也有少数病例的外胚层病变比其他表现出现得更早,提示外胚层病变可能是CCS固有的临床表现而并非继发于腹泻和吸收不良。
实验室检查以贫血、低钾、低钙、低镁血症等电解质紊乱、低蛋白血症(TP和Alb都下降)、胃肠道丢失蛋白增多,肠道乳糖酶缺乏及大便脂肪增多、胃酸减少或缺乏为主。部分患者免疫功能受损,IgG、IgM或IgA下降,补体下降。
内镜检查发现,息肉在全消化道都有分布。绝大多数患者同时有胃和结肠的息肉,个别患者息肉仅侵犯其中之一。小肠中十二指肠息肉数目最多,其次为空肠和近端回肠,到末端回肠息肉数目又有回升。内镜下观察,息肉一般是无蒂的,大小0.2~3.0cm。息肉呈结节状或不规则状,顶部可分叶,息肉黏膜充血水肿。有时可见息肉表面糜烂、溃破和出血。息肉散发或节段性覆盖在胃或结肠黏膜上。息肉之间的黏膜可正常,也可有明显的充血水肿。CCS息肉典型的组织学特点是:①上皮层大多完整,部分腺体有隐窝脓肿;②腺体增生弯曲,部分呈囊性扩张;③上皮和腺体由PAS染色阳性的高大柱状上皮覆盖,扩张的腺管内常充满PAS染色阳性的黏液以及炎症细胞;④固有层炎性水肿,有中至重度的淋巴细胞、浆细胞、嗜酸性粒细胞和中性粒细胞浸润。单纯从形态学方面也很难将CCS的息肉与幼年性息肉病或增生性息肉病的息肉区分开。所以,组织病理学检查也有一定的局限性,诊断CCS必须结合临床。
CCS主要诊断依据为:①成年发病,男性多见;②无家族史;③胃肠道广泛多发息肉;④有外胚层病变如皮肤色素沉着、脱发、指(趾)甲萎缩等;⑤表现为腹泻、腹痛、食欲不振、纳差、体重下降等症状。其中①至④点必须全部具备,缺一不可。
综合以上各点诊断CCS并不困难,但仍需和一些其他肠道息肉病:如黑斑息肉综合征、家族性结肠息肉病、Cowden综合征、幼年型息肉病、家族性结肠息肉病、多发肿瘤(Turcot)综合征、Menetrier病等相鉴别。
此病目前尚无公认的特效疗法,可能“有效”的疗法包括:营养支持、维生素、抗生素、皮质激素、微量元素、抗纤溶治疗、同化激素、抑酸药物、组胺受体拮抗剂、内镜治疗、手术治疗以及联合治疗等。应慎用激素,非激素疗法也能不同程度改善症状,多数学者认为手术治疗不能单纯用来解决症状。足够的支持治疗可有效改善患者的生活质量、降低死亡率,40%~50%的患者长期存活,而50%~60%的患者在确诊后2年内死亡。
(陈学平)
参考文献
1.袁岸龙,寇继光.黑斑息肉综合征患者临床特点调查分析.中华消化杂志,2011,31(6): 417-419.
2. Fotiadis C,Tsekouras DK,Antonakis P,et al.Gardner’s syndrome: A case report and review ofthe literature.World JGastroenterol,2005,11: 5408-5411.
3.黎蕾,张刚,刘婷,等.Gardner综合征的临床影像诊断南方医科大学学报,2009,29(3): 568-569.
4.沈建国,韦菊英,夏惕勤,等.伴有黑棘皮病的胃癌三例.中华内科杂志,2006,45: 326
5. Yeh JS,Munn SE,Plunkett TA,etal.Coexistence of acanthosis nigricans and the sign of Leser2 Trelat in a patientwith gastric adenocarcinoma: a case report and literature review.J Am Acad Dermatol,2000,42: 357-362.
6.武希润,王玲,王琦,等.Cronkhite-Canada综合征国内文献复习.中华内科杂志,2005,44: 387-388.
7.方卫纲,杨爱明,方秀才,等.Cronkhite-Canada综合征的诊治探讨.中华消化内镜杂志,1999,16: 181-182.
8.曹晓沧,周斌,丁娟娟,等.35例中国人Cronkhite-Canada综合征临床分析.中华内科杂志,2007,87: 3130-3132.
来源:《消化科病例分析:入门与提高》
作者:辛永宁 柏愚
页码:365-370
出版:人民卫生出版社
- 评价此内容

 3我要打分
3我要打分


 会员登录
会员登录
近期推荐
- 48岁肥胖男性反酸、胸骨后烧灼…
- 39岁男子腹痛伴便秘2年 别不当…
- 青年女性反复上腹痛伴胃灼热感…
- 58岁女性转移性右下腹疼痛未重…
- 一大叔右上腹隐痛消炎治疗无效…
- 海鲜和烧烤下肚 年轻男子腹痛…
- 42岁男子干农活时呕血两次 问…
- 48岁女性反复右上腹隐痛1年多 …
- 45岁女性上腹痛、间断双下肢水…
- 45岁男子受凉后乏力 自服感冒…

热点文章
- 还没有任何项目!
热门关键词
最新会议
- 2013循证医学和实效研究方法学研讨会
- 欧洲心脏病学会年会
- 世界帕金森病和相关疾病2013年会议
- 英国介入放射学学会2013年第25届年会
- 美国血液学会2013年年会
- 美国癫痫学会2013年第67届年会
- 肥胖学会 2013年年会
- 2013年第9届欧洲抗体会议
- 国际精神病学协会 2013年会议
- 妇科肿瘤2013年第18届大会
- 国际创伤压力研究学会2013年第29届…
- 2013年第4届亚太地区骨质疏松症会议
- 皮肤病协会国际2013年会议
- 世界糖尿病2013年大会
- 2013年国际成瘾性药年会
- 彭晓霞---诊断试验的Meta分析
- 武姗姗---累积Meta分析和TSA分析
- 孙凤---Network Meta分析
- 杨智荣---Cochrane综述实战经验分享
- 杨祖耀---疾病频率资料的Meta分析
友情链接
合作伙伴
Copyright g-medon.com All Rights Reserved 环球医学资讯 未经授权请勿转载!
网络实名:环球医学:京ICP备08004413号-2
关于我们|
我们的服务|版权及责任声明|联系我们
互联网药品信息服务资格证书(京)-经营性-2017-0027
互联网医疗保健信息服务复核同意书 京卫计网审[2015]第0344号
