女36岁,间断发热、四肢乏力、气短、消瘦6个月,加重7天
临床资料
患者女性,36岁,主因间断发热、四肢乏力、气短、消瘦6个月,加重7天于2008年3月30日入院。
患者于入院前6个月无明显诱因出现明显四肢无力,当时无头痛、恶心及发热,给予高压氧治疗2次,症状无改善,10天后出现气短、呼吸急促、声音嘶哑、语音低弱、发热,按“神经根炎”给予激素治疗后症状有所好转,仍反复出现发热(体温在38~39℃)。入院半个月前逐渐出现视物模糊、高热、气短、乏力,精神极差,无力下床行走,饮水时有呛咳,3天前出现神志淡漠、尿失禁。
入院查体:强迫体位,神志清楚,躁动不安,呼吸急促。满月脸,贫血貌。四肢末梢、大腿内侧皮肤花斑,颈部胸前有10cm×10cm瘀斑。构音障碍,右侧鼻唇沟浅,软腭抬举度差,咽反射(+)。左侧肢体肌力4级,右侧肢体肌力3级,双手摸索、强握动作,双侧腹壁反射(+),双侧肱二头、三头肌腱、膝腱及跟腱反射对称(+++),双侧Hoffmann征(+),双侧Babinski征(-),Chaddock征(+)。颈软无抵抗,Kernig征(-)。2008年3月26日头颅MRI(图2.1‐1)左侧中脑、脑桥、丘脑弥漫性肿胀、脱髓鞘改变。
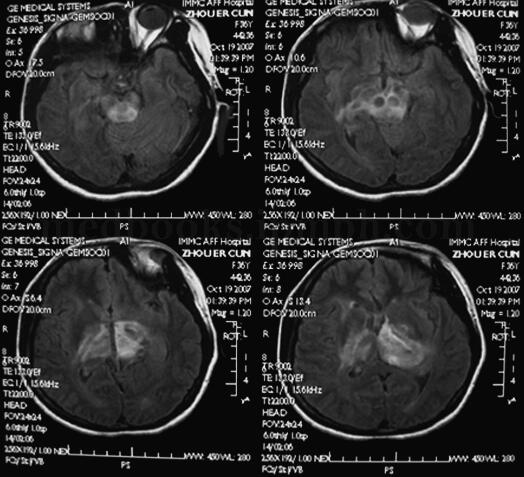
图2﹒1‐1 头颅MRI 左侧中脑、脑桥、丘脑弥漫性肿胀、脱髓鞘改变
入院后给予甲泼尼龙冲击、营养支持、输血及防治并发症等综合治疗,患者病情好转,意识清楚,躁动消失,呼吸频率减慢,贫血纠正。腰穿脑脊液病理诊断结果:涂片中见较多不成熟的淋巴细胞与一些核浆比较大的瘤样细胞,免疫组化呈B细胞表达,考虑淋巴瘤。后确诊为原发性中枢神经系统淋巴瘤而转入血液科,给予ECHOP方案化疗。经治疗患者一般情况可,问答切题,表情自然,可适当走动。
讨论
原发性中枢神经系统淋巴瘤(primary central nervous system lymphoma,PCNSL)最早于1921年由Bailey报道,是指原发于脑实质内,而身体其他部位未发现的淋巴瘤。PCNSL属中高度恶性NHL,其病理类型多为弥漫型大B细胞来源[1],发病率较低,仅占脑内肿瘤的0.3%~1.5%;可发生于各个年龄阶段,以40~50岁居多,男女性比例为2∶1[2]。艾滋病患者、接受脏器移植行免疫抑制治疗者、患有遗传性免疫缺陷病及其他免疫系统疾病者易患本病。
由于PCNSL病灶主要围绕血管周围间隙浸润生长,所以病灶多发生于血管周围间隙较丰富的额顶叶脑白质内近皮质处及胼胝体区近室管膜处[1,3]。一般分为单发和多发两种类型,前者多位于大脑皮质深部,以额叶多见;后者广泛分布于两侧大脑半球、脑干、小脑等区域,可广泛侵犯脑膜[2]。
本病无特征性临床表现,病程较短且进展较快,临床症状常与肿瘤所在的位置有关,主要症状有头痛、呕吐、视乳头水肿和偏瘫,感觉减退,少数患者可有癫痫、精神障碍等。本例患者为青年女性,病程6个月,症状逐渐进展,主要表现为认知功能减退,不自主动作,中枢性过度换气,查体有构音障碍,左侧鼻唇沟变浅,双侧软腭抬举差,咽反射减退,双侧锥体束征阳性。头颅MRI见:沿大脑中线两侧白质为主到脑干、第四脑室周围T1等信号、T2长信号影,结构饱满感。临床表现和体征与多病灶基本符合。
由于对PCNSL认识的不足,早期误诊率较高。影像学检查有一定帮助,虽有相对特征性,如一侧脑叶皮质下类圆形单发病灶或大脑深部不规则多发病灶,很少出现出血、坏死、囊变、钙化等改变,CT与MR多呈均匀强化等[4],但病灶可出现在颅内的任何区域,其发病部位对鉴别诊断的帮助意义不大。与之混淆的病种主要为胶质瘤、脑膜瘤和转移瘤及颅内炎症。所以确定诊断大多依赖术后或立体定位的肿瘤活检,脑脊液细胞学检查也有一定帮助,可发现淋巴瘤细胞或异型淋巴细胞,免疫细胞化学发现部分患者B细胞标记物阳性[5]。本例患者确诊依靠脑脊液病理诊断结果:较多不成熟的淋巴细胞与一些核浆比较大的瘤样细胞,免疫组化呈B细胞表达。
目前治疗一般采用包括手术、放疗和化疗在内的综合治疗,激素也有一定效果。本例患者在早期诊断神经炎时及本次入院后均予应用激素,症状一度缓解,考虑与糖皮质激素可能通过激素受体介导引起淋巴瘤细胞溶解和凋亡,同时也与减轻肿瘤引起的水肿和改善症状有关[6]。但早期应用激素可能使肿瘤的组织学特征改变,因此对需要活检病理结果有一定的影响。
总之,对于临床上有神经精神症状,CT或MRI等检查提示颅内占位性病变者,应除外本病的可能,特别是有免疫缺陷患者更应高度怀疑,并进一步明确诊断。
点评
原发性中枢神经系统淋巴瘤(primarycentralnervous system lymphoma,PCNSL)是指发生于大脑、小脑、脑干、软脑(脊)膜、脊髓和眼,而无全身其他淋巴结和淋巴组织浸润的非霍奇金淋巴瘤(NHL)。这是一种少见的浸润性、多源性恶性肿瘤。
PCNSL的病因和发病机制至今还不清楚,易发于三类免疫缺陷患者:艾滋病(AIDS)、接受器官移植及免疫抑制治疗者、有遗传性免疫缺陷及其他获得性免疫缺陷者。
MRI表现形式多样,有的像肿瘤,有的像炎症,有的与梗死类似,占位效应常不明显,增强后肿瘤实质可呈显著强化。免疫缺陷者的MRI增强后肿瘤可呈非均匀团块状、厚薄不均的环状和形态不规则的斑块强化。而接受过激素治疗的患者MR增强扫描显示肿瘤常无强化,且可缩小。
确诊依赖于病理检查。
PCNSL的部分病例对激素治疗敏感。
来源:《神经科少见病例》
作者:张微微 戚晓昆
参编:张茁 王国强 贺茂林 樊东升 高旭光
页码:81-85
出版:人民卫生出版社
- 评价此内容
- 我要打分

 会员登录
会员登录
近期推荐
- 中年男子胃癌术后5年腹胀难忍 …
- 50岁女性胸痛、咳嗽、痰中带血…
- 中年男子头晕、乏力1月余 原来…
- 48岁女性反复恶心呕吐、意识障…
- PD-1单抗靶向治疗导致垂体炎 …
- 旧疾刚愈新病来袭 颅咽管瘤术…
- 10岁男童发现颅内多发占位 术…
- 34岁男子右腹部疼痛3个月 胸部…
- 中年女性颈背部疼痛1年 除了颈…
- 右肺癌术后7个月发现脑转移 化…

热门关键词
最新会议
- 2013循证医学和实效研究方法学研讨会
- 欧洲心脏病学会年会
- 世界帕金森病和相关疾病2013年会议
- 英国介入放射学学会2013年第25届年会
- 美国血液学会2013年年会
- 美国癫痫学会2013年第67届年会
- 肥胖学会 2013年年会
- 2013年第9届欧洲抗体会议
- 国际精神病学协会 2013年会议
- 妇科肿瘤2013年第18届大会
- 国际创伤压力研究学会2013年第29届…
- 2013年第4届亚太地区骨质疏松症会议
- 皮肤病协会国际2013年会议
- 世界糖尿病2013年大会
- 2013年国际成瘾性药年会
- 彭晓霞---诊断试验的Meta分析
- 武姗姗---累积Meta分析和TSA分析
- 孙凤---Network Meta分析
- 杨智荣---Cochrane综述实战经验分享
- 杨祖耀---疾病频率资料的Meta分析
友情链接
合作伙伴
Copyright g-medon.com All Rights Reserved 环球医学资讯 未经授权请勿转载!
网络实名:环球医学:京ICP备08004413号-2
关于我们|
我们的服务|版权及责任声明|联系我们
互联网药品信息服务资格证书(京)-经营性-2017-0027
互联网医疗保健信息服务复核同意书 京卫计网审[2015]第0344号
