一例PSEN1基因突变的早发型阿尔茨海默病病例解析
【导读】阿尔茨海默病(Alzheimer’s disease,AD)是一种以进行性认知功能障碍为特征的中枢神经退行性病变,是最常见的痴呆类型。AD根据发病年龄可分为65岁之前发病的早发型阿尔茨海默病(early onset Alzheimer’s disease,EOAD),约占AD病例的5%~10%,和65岁以后(含65岁)发病的晚发型阿尔茨海默病(late onset Alzheimer’s disease,LOAD),LOAD约占AD病例的90%~95%。此外,按有无AD家族史,又分为散发性AD(sporadic Alzheimer’s disease,SAD)及具有明显家族遗传的家族遗传性AD(familial Alzheimer’s disease,FAD)。FAD约占AD病例的0.6%,EOAD病例的11%。AD病因至今仍不清楚,除了神经系统老化外,家族遗传史是其主要危险因素,目前已经发现200多种基因位点的突变与FAD的发病相关,与FAD遗传有关的编码蛋白基因最常见的为:淀粉样前体蛋白(amyloid precursor protein,APP)基因、早老素1(presenilin 1,PSEN1)基因、早老素2(presenilin 2,PSEN2)基因。
【病例简介】
1.主诉
记忆力进行性减退2年。
2.现病史
患者男性,41岁,小学二年级文化,蔬菜经营者。家属诉患者2年前开始,无明显诱因下逐渐出现记忆减退,表现为前讲后忘,反复询问相同的问题,无法找到个人物品。患者以售卖蔬菜为生,记忆下降逐渐影响正常工作,约1年前开始反应明显迟钝,无法独立完成进货、售卖等一系列工作,需要有人帮助或干脆休息在家。约半年前,患者行动变慢,行走欠稳,偶有跌倒,无摔伤。病程中伴有不自主流涎。家属否认患者有妄想、幻觉和不自主行为等明显精神症状。起病来,患者神志清,精神状态一般,睡眠及胃纳好。
3.既往史
既往体健,否认糖尿病、高血压病史,否认外伤,否认手术,否认重大脏器疾病史。
4.个人史
已婚,育1子1女,配偶体健。无烟酒嗜好,无冶游史。
5.家族史
患者的外婆、母亲、二姨、三舅及姐姐均有记忆力减退情况,且在病程中均出现记忆力减退进行性加重,并逐渐出现四肢僵硬、行动变缓,都在40多岁去世,家系图谱见图1。
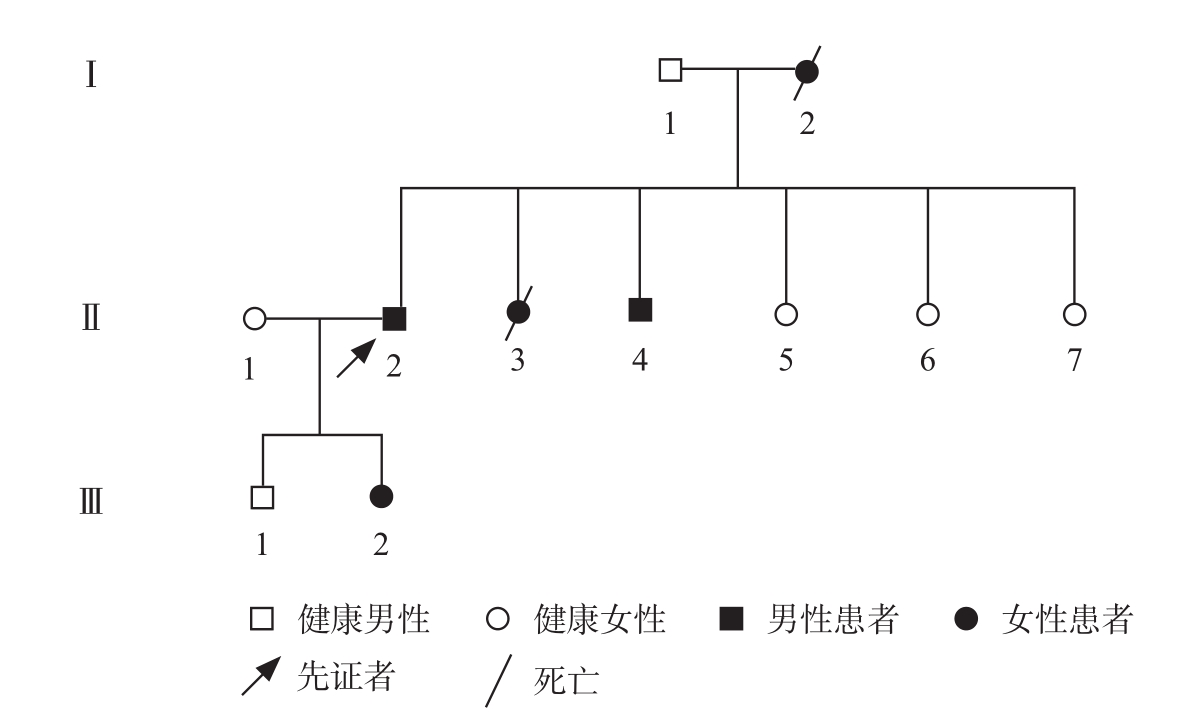
图1 患者家系图谱
6.查体
(1)神志清,言语欠流利,眼球活动尚可,眉心征(+),双侧掌颏反射(+),四肢肌力5级,肌张力正常,双侧轮替活动(+),闭目难立试验(-),直线行走尚能完成。双上肢腱反射对称(++),双下肢腱反射对称(+++),双侧病理征(-)。
(2)精神状态:衣饰尚整齐,被动接触,对答缓慢,内容切题。承认病史中家属反映的情况,称自己“大脑不如以前,可能是生病了”“以前能卖菜,现在算不过来”。知道家里有人“病和自己差不多,死得比较早”,现在自己“快和他们一样了,走路也不行了”,诉自己现在只能待在家里。未发现思维逻辑结构障碍,未见幻觉、妄想,未见怪异行为。接触交谈显得很平静,未发现明显焦虑、抑郁情绪。表示愿意配合检查和治疗。
(3)认知:简易精神状态检查(MMSE)总分16分,定向力6/10分,即刻记忆1/3分,延迟回忆3/3分,注意力和计算力1/5分,语言及运用能力4/8分,复杂命令(画图)1/1分。
7.辅助检查
(1)头颅MRI示:双侧侧脑室体旁及额顶叶多发小缺血灶,未见明显海马萎缩(图2)。
(2)脑电图示:弥漫性慢波,中度异常脑电图。
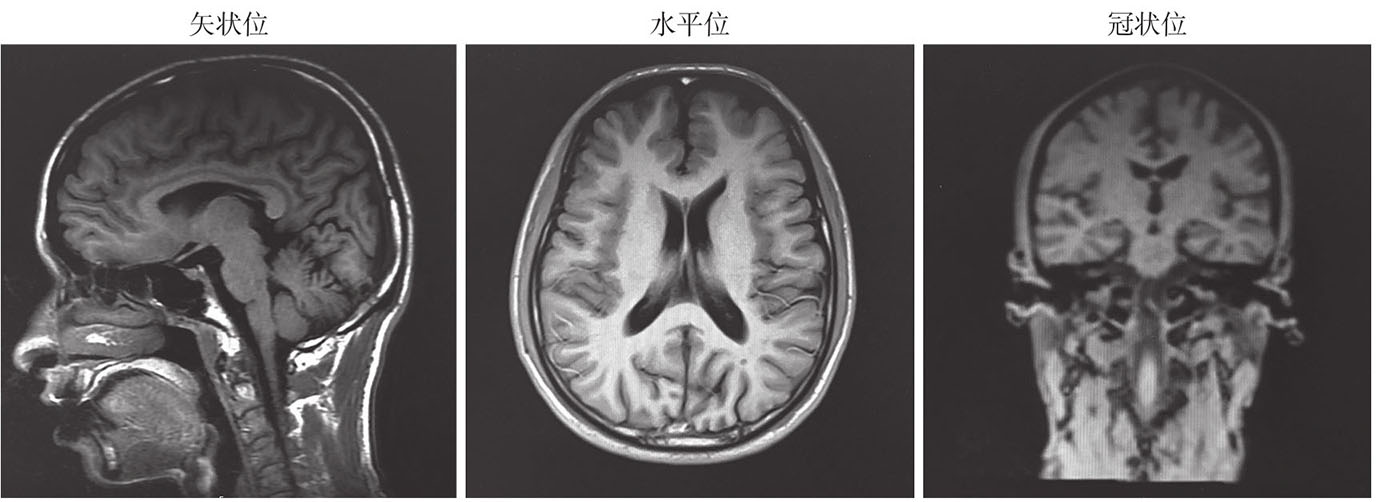
图2 患者头颅磁共振表现
(3)基因检测:患者PSEN1基因存在P284L的杂合突变(8号外显子,编码链错义突变,图3)。载脂蛋白E基因(APOE):ε3/ε3。对患者亲属的该基因位点进行验证发现其弟弟和女儿也存在同样的突变。
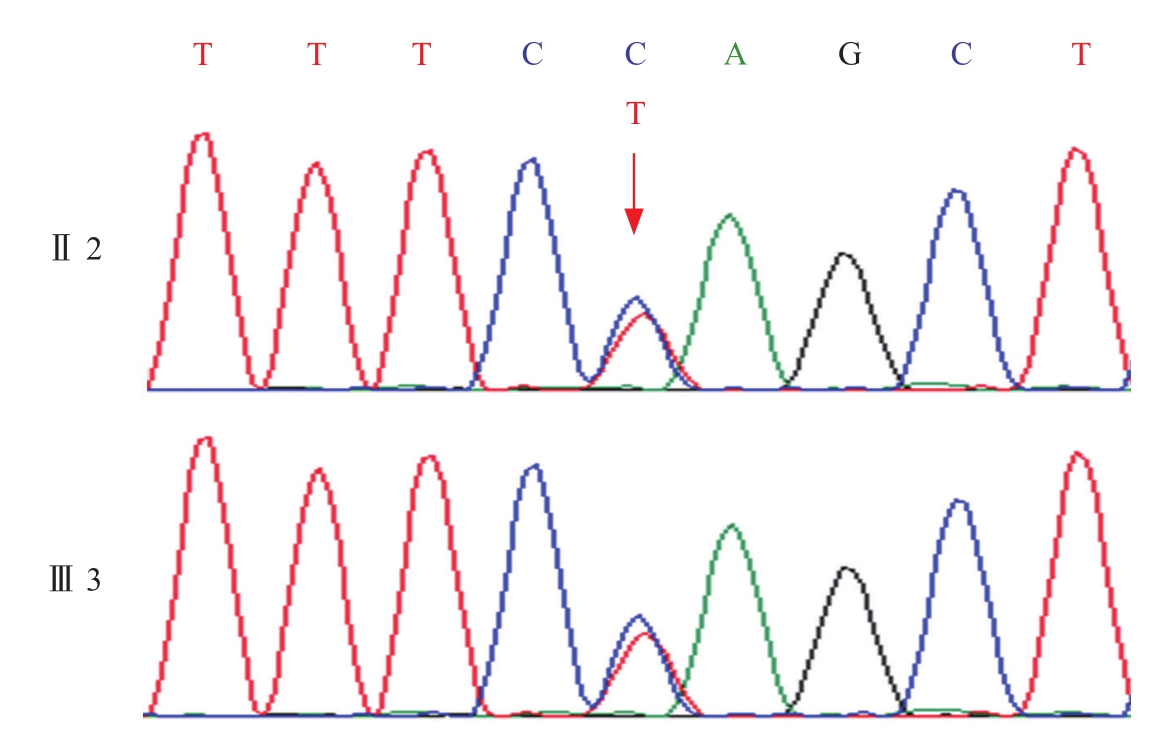
图3 基因测序结果
(4)11C-PiB-PET/CT显像:
未见明显淀粉样蛋白沉积(图4)。
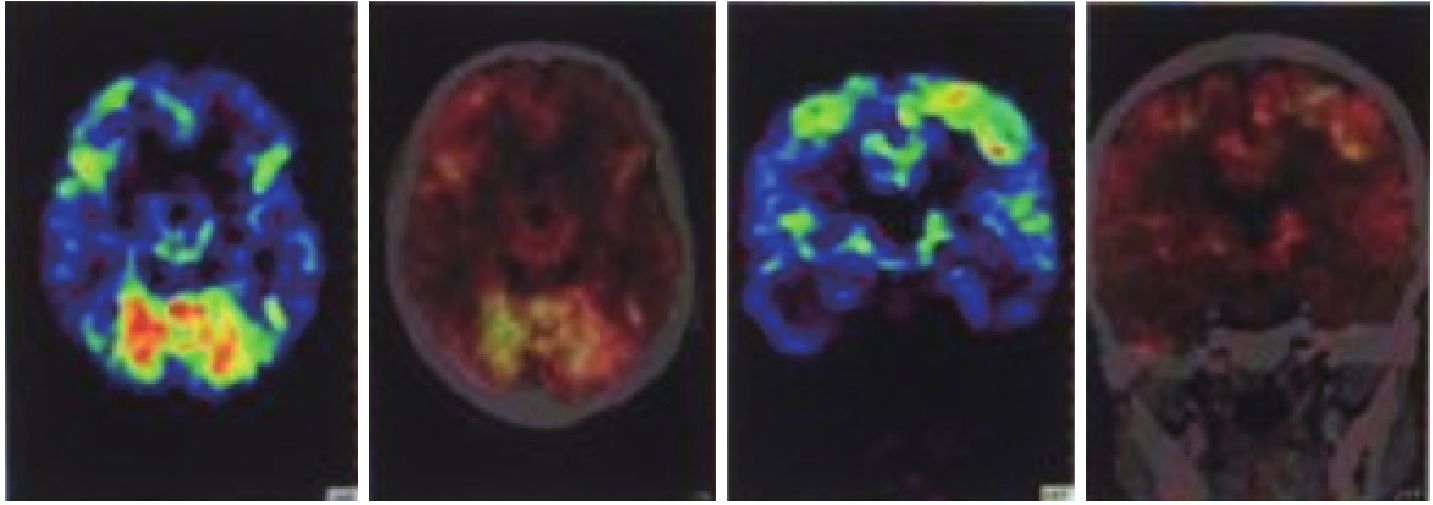
图4 患者11C-PiB-PET/CT表现
8.入院诊断
认知障碍查因。
【临床分析与决策】
患者中青年起病,以记忆等认知功能下降为主要表现,并进行性加重,认知障碍累及多个认知域,伴轻度运动功能下降,无明显精神行为改变,无血管性、感染性和肿瘤疾病史。主要阳性体征为:言语欠流利,眉心征(+),双侧掌颏反射(+),双下肢腱反射(+++)。MMSE为16分(患者文化水平小学二年级);脑电图示双侧慢波增多;头颅MRI示双侧侧脑室体旁及额顶叶多发小缺血灶,双侧海马无萎缩。
由于患者发病年龄早,缓慢起病,无症状波动或复发缓解等特点,头颅MRI未提示明确结构性病变和异常信号,故不考虑脑血管病、脱髓鞘病、中毒代谢类和肿瘤类疾病。根据患者的发病年龄和临床表现,进入早发型痴呆的神经退行性疾病谱进行鉴别。患者以认知障碍表现首发并且相对突出,运动迟缓较轻,有上运动神经元受累,共济受累的线索,无锥体外系受累体征。因此,在痴呆相关的神经退行性疾病谱中,首先排除以帕金森病为代表的突触核蛋白病(包括帕金森病、帕金森病痴呆、路易体痴呆、多系统萎缩),需要考虑tau蛋白病和β淀粉样蛋白沉积病,且患者无进行性核上性麻痹(progressive supranuclear palsy,PSP)的运动障碍表现。
患者有明确的家族史,需首先考虑FAD,为进一步明确诊断,除常规痴呆鉴别诊断检查外,应进行相应的分子遗传学检查,淀粉样蛋白功能影像或脑脊液(CSF)Aβ42,tau蛋白和p-tau蛋白检测。完善检查后作出如下诊断。
1.定位诊断
患者记忆力减退,反应迟钝,记忆力、计算力、推理和判断能力,执行功能和处理复杂任务的能力均下降。MMSE总分16分,小学两年级文化水平(定向力6/10分,即刻记忆1/3分,延迟回忆3/3分,注意力和计算力1/5分,语言及运用能力4/8分,画图1/1分)。可定位于海马以及额颞叶相关高级皮质功能异常。
2.定性诊断
患者中青年起病,病程呈慢性渐进性发展。主要症状表现记忆力及其他多个认知域损害,表现为记忆减退,反应迟钝,计算力、推理和判断能力,执行功能和处理复杂任务的能力均下降,无明显精神行为症状;且患者逐渐出现行动变慢,行走欠稳。定性为神经退行性疾病,虽然患者头颅MRI检查未见明显海马萎缩。11C-PiB-PET/CT显像:未见明显淀粉样蛋白沉积,并不支持AD诊断。但患者基因二代测序结果提示PSEN1基因存在P284L的杂合突变(8号外显子,编码链错义突变)。该突变在患者家系的其他亲属中得到了验证,先证者的弟弟(Ⅱ4)及先证者的女儿(Ⅲ2)均携带该突变。故明确诊断为:PSEN1基因突变的早发型阿尔茨海默病。
3.鉴别诊断
(1)皮质基底节变性(corticobasal degeneration,CBD):CBD发病通常在60~80岁发病,首发症状最常见在61~64岁。经典的CBD表现为进行性不对称性运动障碍,从单肢开始,出现少动、强直、局灶肌阵挛、观念运动性失用,异己手等。认知症状的表现中,注意、执行速度和认知控制缺陷类似于AD,但不同的是,CBD的语词记忆优于AD,运动技能应用差于AD,并且常常表现抑郁情绪。CBD的其他表现包括:腱反射亢进,病理征阳性,眼球及眼睑活动障碍,构音障碍和吞咽障碍。
本患者符合CBD的某些症候群,如认知障碍、言语欠流利、锥体系受累表现。但CBD的起病年龄相对更晚,且存在突出的运动障碍,多为不对称性,进行性加重。而且,CBD的认知障碍集中体现在失用、失语、行为改变和执行功能障碍。本患者运动障碍不明显,认知方面的MMSE失分点并不包括语言及行为运用方面,故暂不考虑此诊断。
(2)额颞叶痴呆(frontotemporal dementia,FTD):FTD是额颞叶变性(frontotemporal lobar degeneration,FTLD)的主要亚型之一。FTLD是一组以额颞叶萎缩为主要病理表现,以进行性精神行为异常、执行功能障碍和语言损害为主要临床特征的痴呆症候群。根据临床特征,病理异常包括tau蛋白、TDP-43,FUS等异常蛋白沉积,突变基因包括MAPT、GRN、C9ORF72等。目前研究发现临床表现与实际病理异常或基因突变的结果并不一一对应。就临床表现而已,本患者的起病年龄符合FTD常见年龄段,在40~75岁段,同时也有遗传相关的家族史。但患者的临床表现与典型FTD不同,本患者并非以精神行为异常起病,且病程中语言理解和语言运用功能障碍并不突出,影像学未提示明显额颞叶萎缩的表现,因此从临床上不考虑FTD诊断。
(3)脑淀粉样血管病(cerebral amyloid angiopathy,CAA):CAA多为散发,但也可表现为家族性综合征。CAA血管淀粉样蛋白沉积与AD类似,也是39~43个氨基酸的β淀粉样多肽,但大多沉积于颅内血管,而非脑实质,且以后半球为主。常见的突变基因包括APP和APOE,其中APOE ε2和ε4是风险基因,ε3是非风险基因。CAA患者的认知下降主要表现在速度感知能力和情景记忆,一般认为和小血管多发微出血和微梗死相关。CAA患者由于存在小血管病变,常常合并脑血管事件,包括脑叶出血、短暂性脑缺血发作(transient ischemic attack,TIA)、微出血等。本患者有认知下降及相关家族史,需考虑CAA,但起病年龄较常见CAA早(CAA多于60岁以后起病,几乎没有50岁以下的病例报道),且在2年左右的病程中无明显脑血管意外发生。虽然目前临床上不支持CAA,但由于CAA和AD可以叠加出现并导致认知症状更为严重,故需要通过磁共振磁敏感扫描、淀粉样蛋白成像和基因筛查来进一步明确。
【诊断】
PSEN1基因突变的早发型阿尔茨海默病
【诊治过程】
患者小学二年级文化,MMSE 16分,基本生活功能大多保留,故先给予胆碱酯酶抑制剂治疗(卡巴拉汀,起始剂量为1.5mg,每日2次口服;患者服用2周以后对此剂量耐受良好,剂量增至3mg,每日2次口服),并随访。
【预后及随访】
1年后患者病情加重加用美金刚治疗。
2017年对患者进行随访。患者记忆力、语言理解力、定向力均呈进行性下降,肌力、肌张力均无变化,且无癫痫、肌阵挛、痉挛性瘫痪、假性延髓性麻痹、精神症状等。复测MMSE量表总分7分(定向力2/10分,即刻记忆0/3分,延迟回忆0/3分,注意力和计算力1/5分,语言及运用能力3/8分,画图1/1分)。
2017年复查,11C-PiB-PET/CT显像,提示双侧大脑(额叶,后扣带回,顶叶,枕叶,颞叶)皮质广泛淀粉样蛋白沉积。
2019年随访,MMSE 7分,淀粉样蛋白PET/MRI提示双侧大脑(额叶、后扣带回、顶叶、枕叶、颞叶)皮质广泛淀粉样蛋白沉积。
【讨论】
阿尔茨海默病(Alzheimer’s disease,AD)是多种因素共同作用、具有较高遗传异质性的常见神经退行性疾病。遗传因素被确认在AD特别是在EOAD的发病过程中扮演了重要角色。EOAD与LOAD两型均可有遗传因素参与,EOAD通常为常染色体显性遗传,而LOAD以散发多见。近年来,AD相关的遗传学和分子生物学研究取得了明显进展,目前已发现至少200个基因位点与AD的发病相关,其中最常见的是位于14q24.3的早老素1(presenilin-1,PSEN1)基因、位于lq31-42的早老素2(presenilin-2,PSEN2)基因和位于21ql 1.2-22上的淀粉样前体蛋白(amyloid precursor protein,APP)基因。占AD 90%的SAD患者,影响其发病的遗传因素多为携带APOE4等位基因或上述AD风险基因。研究表明PSEN1、PSEN2、APP三个致病基因与FAD发病密切相关。约80%的EOFAD患者中携带这三种致病基因突变。不同突变形式的EOFAD患者有不同的临床表型,同一种基因突变的家系里不同的个体也有不同的临床表型。
EOAD对比LOAD有更显著的遗传易感性(包括常染色体显性遗传易感性,以及常见的多基因遗传易感性)。EOAD与LOAD(不包括APOE)相比,前者在诊断前有较长的病程(约1.6年),但临床总体进展速度更快,死亡风险高于LOAD的患者,很多EOAD的患者在40~64岁死亡。EOFAD患者较散发性EOAD患者平均发病年龄更早(约14年),病程更长(约2年)、MMSE评分更低,可伴头痛、肌阵挛等症状,发病后5~12年甚至伴发癫痫,散发性EOAD患者更易早期出现语言功能损伤、视空间缺损、失用症、日常行为功能障碍等。
EOFAD的患者可出现认知及精神行为外的临床征象,如:头痛、肌阵挛、癫痫、步态异常、假性延髓性麻痹和反射亢进等。广泛皮质萎缩尤其是顶叶皮质和正常海马或轻度萎缩是EOAD患者的影像学结构改变特征。功能磁共振成像(fMRI)检查进一步发现与LOAD的患者相比,EOAD的患者与海马连接的皮质区域的功能变化较小,支持了上述结构影像学表现。
FAD研究表明,不同的突变基因,甚至相同基因上的不同突变位点造成的细胞病理损伤并不完全相同,从而其临床表型也不同。在FAD家系中,PSEN1基因突变是最常见、突变位点报道最多的基因,APP基因突变次之,PSEN2基因突变最少。PSEN1基因突变的患者发病年龄较早,易出现肌阵挛、癫痫、锥体外系症状及精神行为异常,而语言功能损伤、小脑性共济失调及痉挛性下肢轻瘫则比较少见。不同APP基因突变类型的临床特征不尽相同,存在异质性。APP突变的患者除表现为记忆力进行性下降以外,更频繁地表现为攻击性。APP基因重复突变的患者可出现失用。PSEN2基因突变患者发病年龄相对PSEN1、APP基因突变患者较晚,部分患者无阳性家族史,易诊断成散发型AD,临床表现除AD的主要特征外,伴有其他症状如锥体系、锥体外系症状、定向障碍、帕金森症候群、肌阵挛等症状。
【专家点评】
讨论问题1:哪些AD需要遗传学检测?常见基因异常有哪些?
从临床起病时间划分,65岁以前起病的被称为早发型痴呆。英国2003年的一项流行病学调查表明,早发型痴呆中34%为早发型AD(EOAD),所占比重最大(18%为血管性痴呆,12%为FTD,7%为路易体痴呆,10%为酒精相关性痴呆,19%是其他)。EOAD虽然只占所有AD的10%,但通常有很强的遗传背景,并且表现为常染色体显性遗传。在这些家庭中,几乎每一代都有人患病,起病年龄在30~60岁。值得注意的是,由于部分患病家庭成员“不明原因”早逝,这可能会影响医师对家族史的判断,因此需要仔细询问。这部分患者最需要遗传学检测,并与其他神经遗传变性疾病相鉴别。
由于基因检测昂贵,并且目前尚无针对性基因治疗,因此要严格根据临床需要选择性地进行:在EOAD中,60%~70%的EOAD与以下三个基因的致病突变有关:位于1号染色体的PSEN2,14号染色体的PSEN1和21号染色体的APP。这些致病突变的外显率(penetrance)极高,携带者100%会发病,因此需要临床高度重视。在三种突变中,PSEN1突变的发病年龄最早,中位年龄43岁。
在晚发型AD(LOAD)中,如果本人尚未发病,而其一级亲属和兄弟姐妹发生AD,则本人发生AD风险较正常情况提高10%~30%。但对如果一级亲属发生AD的年龄高于85岁,则此AD为遗传性的可能性几乎为零。LOAD尚缺乏比较明确的致病基因,并且环境因素和表观遗传学因素对疾病发生发展的影响更大。目前唯一具有强循证依据,并且能够被称为LOAD危险因素的基因是APOE ε4。
讨论问题2:本例中,PSEN1突变类型与AD发病的关系是怎么样的,能否解释患者的临床症状?
PSEN1突变所致家族性早发型AD呈常染色体显性遗传,其突变所致家族性早发型AD约占18%~50%,目前已有超过160种的PSEN1的致病突变已经报道,以错义突变为主。PSEN1是γ分泌酶复合体的重要组成部分,其突变导致β淀粉样蛋白Aβ42的增多,导致在AD患者脑组织中淀粉样斑块的沉积。
2002年,PSEN1基因P284L突变(8号外显子,编码链错义突变)在日本首次报道(也是目前能查询到的唯一1例公开报道),其临床特点是除了表现为认知障碍外,随着疾病的进展可伴有神经系统的其他症状,如癫痫、肌阵挛、痉挛性瘫痪、假性延髓性麻痹等症状,以痉挛性瘫痪和强直最为常见。病理上以棉絮样斑(CWPs)广泛分布于整个中枢神经系统为特征,核心斑块主要见于海马和小脑,胶质炎性反应非常轻微。海马、蓝斑和黑质的神经元减少最为明显,其次是皮质和基底节。β淀粉样蛋白沉积于血管壁及周围区域,伴许多微梗死和微出血。笔者采用电镜对棉絮样斑(CWPs)进行成分分析,发现其主要成分是突触成分中的电子致密囊泡,淀粉样纤维极少。P284L突变与此病理改变的具体机制尚待研究。
本例患者表现认知相关高级皮质及皮质下联络纤维受累,同时伴有锥体系部分受累。同时,患者的一级家属病程中均出现四肢僵硬,行动变缓,且都在40多岁去世。因此,虽然目前没有获取脑活检的条件来证实患者的PSEN1突变与病理的关系,但患者的临床表现符合已知病例报道,并且家系中基因突变和临床表现基本一致。故从临床上判断患者家系中PSEN1突变导致一系列临床症状。
讨论问题3:如何解读此病例的PET-PiB结果和MRI结果?
碳11标记的匹兹堡复合物B(PiB)是一种硫黄素T的放射性同位素(11C-PiB),可以在PET扫描中显影神经组织中的β淀粉样蛋白(Aβ)。11C-PiB特异性地与Aβ纤维和含有Aβ的不溶性斑块结合,但不能和可溶性或非纤维Aβ结合(除非含量极大)。同时,PiB也不与神经原纤维缠结(NFTs)结合。在淀粉样蛋白所致疾病CAA及AD患者中,PET-PiB均能够显示异常Aβ沉积。总体而言,AD沉积最明显,CAA其次,健康对照最低。其中,AD的额叶和颞叶沉积相对量高于CAA,而CAA的枕叶区沉积相对量高于AD。
本患者的PET-PiB结果未见异常淀粉样蛋白沉积,可能有两个原因。第一,PSEN1突变增加了Aβ的清除率比例(fractional turnover rate,FTR),更多的Aβ沉积在斑块中,CSF中的Aβ含量减少。PSEN1动物模型证实,Aβ不可逆的FTR增加在斑块形成前就已经发生,此时进行PET-PiB检查不能100%得到阳性结果。第二,目前的PiB分析方法默认小脑为Aβ阴性区域,故大多以小脑作为参考区,将大脑皮质感兴趣区的PET 标准摄取值(SUV)与小脑的SUV值做比较,得到大脑皮质标准摄取值比值(SUVR)。SUVR是评定PET阴性和阳性的依据。与本例相同基因突变的病理结果显示,CWP核心斑块主要见于海马和小脑。因此,如果以小脑作为参考区计算SUVR,可能得出假阴性的结果。
本患者虽然有明显的认知障碍表现,但头颅MRI提示双侧侧脑室体旁及额顶叶小缺血灶,双侧海马无萎缩。这与典型的AD又有差异。根据病例报道中的病理结果,PSEN1基因P284L突变病理上发现β淀粉样蛋白沉积于血管壁及周围区域,伴许多微梗死和微出血。因此,在疾病早期,认知功能下降可能首先归结于微血管病变,而非结构性萎缩。此外,遗传型AD协作数据库(DIAN)曾做出统计分析结果显示,常染色体显性遗传AD患者的T2加权像中的白质高信号是独立于微血管病变,可作为AD的独立核心特征。总之,结构是否萎缩并不与认知功能平行,仅仅能作为临床诊断和判断疾病程度的辅助指标之一。
参考文献
[1]SUDHIR K.Memantine Pharmacological properties and clinical Uses[J].Neural India,2004,52(3): 1307-1309.
[2]BIRD T D.Genetic aspects of Alzheimer disease[J]. Genet Med,2008,10(4):231-239.
[3]BEKRIS I M,YU C E,BIRD T D,et al.Genetics of Alzheimer disease[J].Geriatr Psychiatry Neurol,2010,23(4):213- 227.
[4]GENIN E, HANNEQUIN D, WALLON D, et al. APOE and Alzheimer disease: a major gene with semidominant inheritance[J]. Molecular Psychiatry,2011, 16(9):903.
[5]LAMBERT J C, IBRAHIM-VERBAAS C A, HAROLD D, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease[J]. Nature Genetics,2013,45(12):1452-1458.
[6]GERRITSEN A A, BAKKER C, VERHEY F R, et al. Prevalence of comorbidity in patients with young-onset Alzheimer disease compared with late-onset: a comparative cohort study[J]. Am Med Dir Assoc,2016,17(4):318–323.
[7]WATTMO C, WALLIN Å K. Early- versus late-onset Alzheimer’s disease in clinical practice: cognitive and global outcomes over 3 years[J]. Alzheimers Res Ther,2017,9(1):70.
[8]CHANG K J, HONG C H, LEE K S, et al. Mortality risk after diagnosis of early-onset Alzheimer’s disease versus late-onset Alzheimer’s disease: a propensity score matching analysis[J]. Alzheimers Dis,2017,56(4):1341-1348.
[9]CHEN Y, SILLAIRE A R, DALLONGEVILLE J, et al. Low prevalence and clinical effect of vascular risk factors in early-onset Alzheimer’s disease[J]. Alzheimers Dis,2017,60(3):1045–1054.
[10]DICKERSON B C, BRICKHOUSE M, MCGINNIS S, et al. Alzheimer’s disease: the influence of age on clinical heterogeneity through the human brain connectome[J]. Alzheimers Dement (Amst),2016,6: 122–135.
[11]SHEA Y F,CHU L W,CHANC A O K,et al. A systematic review of familial Alzheimer’s disease:Differences in presentation of clinical feature among three mutated genes and potential ethnic diffrences[J].Formosan Medical association,2016,115(2):67-75.
[12]GUERREIRO R J,BAQUERO M,BLESA R et al. Genetic screening of Alzheimer’s disease genes in lberian and African samples yields novel mutations in presenilins and APP[J]. Neurobiol Aging, 2010,31(5):725-731.
知识来源
人卫知识数字服务体系
- 评价此内容

 3我要打分
3我要打分



 会员登录
会员登录
近期推荐
- 全外显子测序和脑脊液AD标志物…
- 48岁男子下肢麻木、视物模糊 …
- 79岁女性记忆力减退且性格改变…
- 35岁女性反复发作性头痛16年 …
- 51岁男性夜间起床时突发左侧肢…
- 68岁男子与他人吵架后半小时突…
- 62岁男性突发垂体瘤卒中 手术…
- 49岁男子因坠落致颅脑损伤 多…
- 花季少女多饮多尿增重停经 MRI…
- 37岁女性浑身无力且恶化很快 …

热点文章
- 还没有任何项目!
热门关键词
最新会议
- 2013循证医学和实效研究方法学研讨会
- 欧洲心脏病学会年会
- 世界帕金森病和相关疾病2013年会议
- 英国介入放射学学会2013年第25届年会
- 美国血液学会2013年年会
- 美国癫痫学会2013年第67届年会
- 肥胖学会 2013年年会
- 2013年第9届欧洲抗体会议
- 国际精神病学协会 2013年会议
- 妇科肿瘤2013年第18届大会
- 国际创伤压力研究学会2013年第29届…
- 2013年第4届亚太地区骨质疏松症会议
- 皮肤病协会国际2013年会议
- 世界糖尿病2013年大会
- 2013年国际成瘾性药年会
- 彭晓霞---诊断试验的Meta分析
- 武姗姗---累积Meta分析和TSA分析
- 孙凤---Network Meta分析
- 杨智荣---Cochrane综述实战经验分享
- 杨祖耀---疾病频率资料的Meta分析
友情链接
合作伙伴
Copyright g-medon.com All Rights Reserved 环球医学资讯 未经授权请勿转载!
网络实名:环球医学:京ICP备08004413号-2
关于我们|
我们的服务|版权及责任声明|联系我们
互联网药品信息服务资格证书(京)-经营性-2017-0027
互联网医疗保健信息服务复核同意书 京卫计网审[2015]第0344号
